Molecular Window
Molecular manipulation
When the file is opened, a new window appears and the corresponding molecule is displayed in 3D as a ball-and-stick model. You can change the background color and other settings in the “Preferences” dialog. For details, please read “Preferences” in this manual.
Molecular Rotation Operations
The displayed molecular structure can be rotated using the mouse or trackpad. Trackpad operation may vary depending on the model of your computer. Please refer to your computer's manual for the operation method that corresponds to the following mouse operations.
Hold down the left mouse button and drag to rotate the molecule.
You can rotate a molecule around a perpendicular line to the window by holding down the Option or Alt and Shift keys on the keyboard at the same time and clicking and dragging the left mouse button.
Zooming
If your mouse has a scroll wheel, you can rotate the wheel to enlarge or shrink the molecules.
Parallel shift operation
Hold down the Option or Alt key on the keyboard while holding down the left mouse button and dragging to translate the molecule.
Cancel Operation
To cancel all rotation and translation operations and return to the initial state, click the “Init”. icon on the controller (see below).
Display Format
When you open a molecule file, a new window will appear and the molecule will be displayed in a ball-and-stick 3D model. The background color, etc. can be changed in the “Preferences” dialog. For details, refer to “Preferences” in this manual. In addition, the name of the file is displayed in the window's title bar.
The display format can also be changed to “CPK” or “Wire Frame” using the buttons in the [Molecule Box] or the View menu. You can also use the Symbol and Label buttons or the View menu to display labels overlaid on atoms.
Partial change of display format
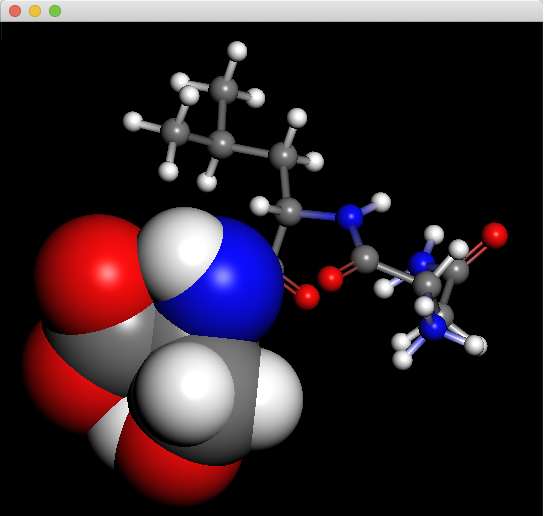
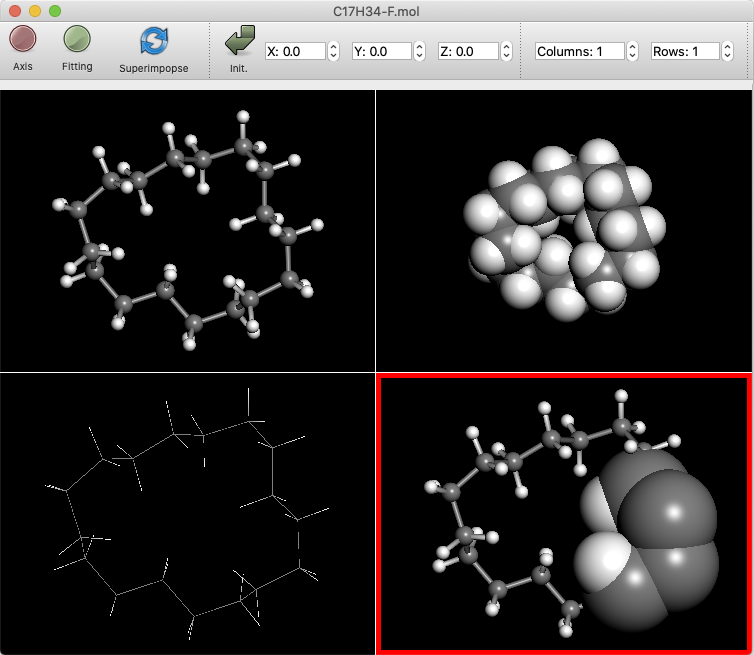
The Molecular Display can also display a combination of “CPK”, “Ball & Stick”, and “Wire Frame”.
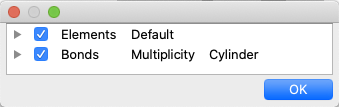
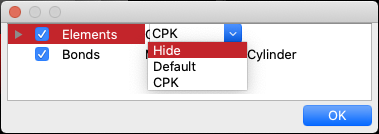
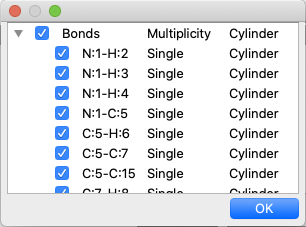
By right-clicking the mouse in the molecule window, a context menu will appear. Selecting “Setup Display” from the menu will display the “Setup Display” dialog, which shows the components of the molecule. Unchecking the checkboxes to the left of each item will hide the corresponding item from being displayed. You can also change the display method by double-clicking on the attribute next to the item name, either by typing it in directly or by selecting from the options. For “Elements”, you can select from the following or directly enter a value between 0.0 and 1.0.
- Hide
- Default
- CPK
These correspond to atomic radius ratios of 0.0, 0.2, and 1.0, respectively. If you wish to set a ratio other than these, you can do so by entering a numerical value directly (e.g., 0.5). Changing the setting next to “Elements” will modify the selected area to the same setting. You can also click on the leftmost triangle icon to display a group of atoms and set them individually.
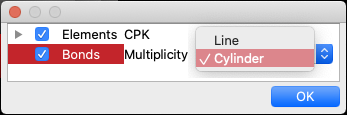
For “Bonds”, choose from the following options:
- Line
- Cylinder
As for bond, it is not possible to adjust the thickness of the cylinder by entering a numerical value. The line width in “Line” view can be set in “Preferences”.
If you wish to change the display of individual bonds, you can click on the left-most triangle icon to display the bonds individually and adjust them as in “Elements”.
The molecular display shown below combines the CPK display with the ball&stick display.

Selection operation
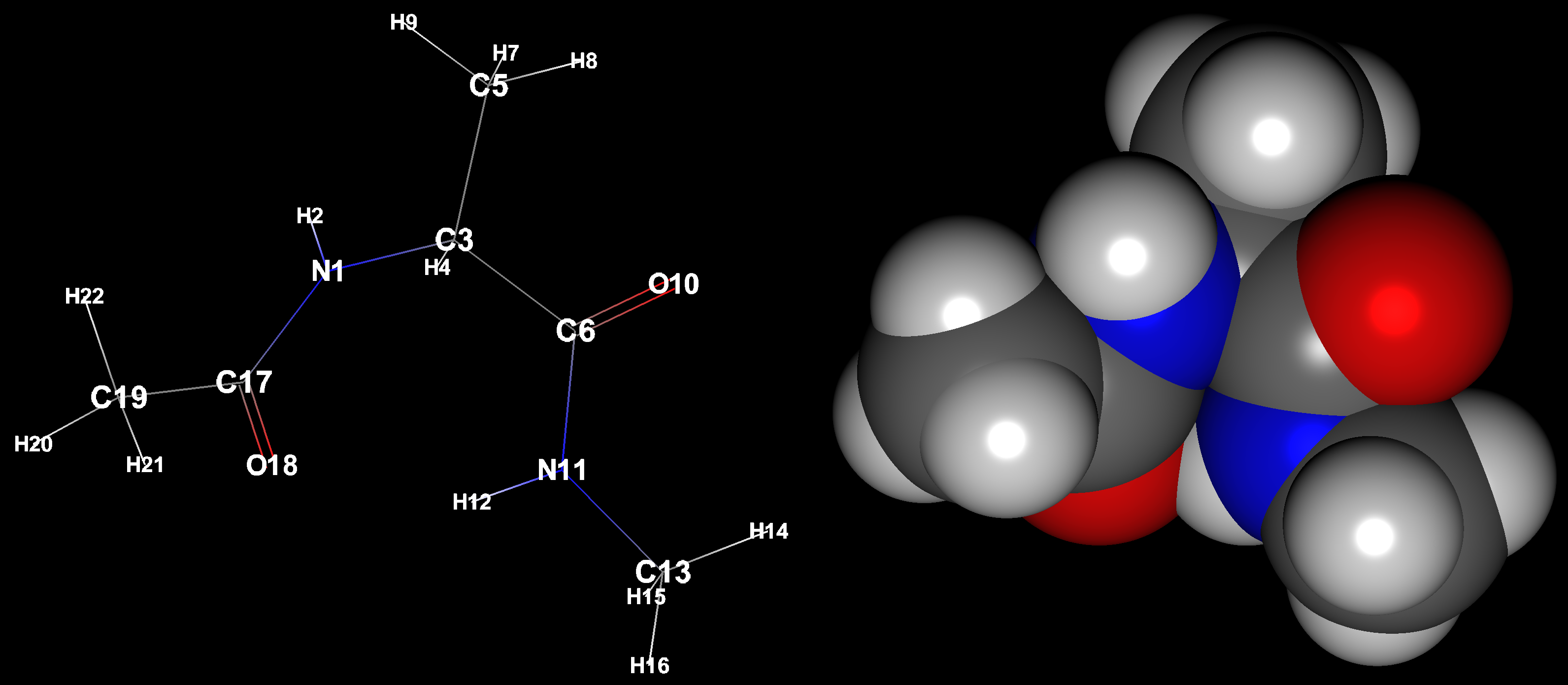
By clicking on an atom with the mouse, you can select that atom. When selected, other areas are darkened and the selection is highlighted. Once selected, the contents of the “Object Inspector” dialog will be updated to show information about the selected atom. In addition, the bond lengths, bond angles, and dihedral angles containing the selected atoms are also displayed. By clicking the angle data displayed here with the mouse, the corresponding data will be labeled in the molecule window. The figure shows an example in which one atom is selected and one of the Angle Lists in the displayed Angle List is selected to display the angle labels.
After selecting an atom, you can hold down the Shift key on the keyboard and click on the atom to add a selection. At the same time, the bond length will be labeled if the number of atoms selected is 2, the bond angle if 3, and the dihedral angle if 4. If more than that, nothing is displayed.
By double-clicking on an atom, you can select the entire molecule that contains that atom. This is useful when you have opened a file containing multiple molecules and wish to select only a specific molecule. The figure below shows a double-click on an atom that selects the molecule containing that atom.
If you want to select the whole molecule while a part of it is selected, click on an empty area, such as an atom.

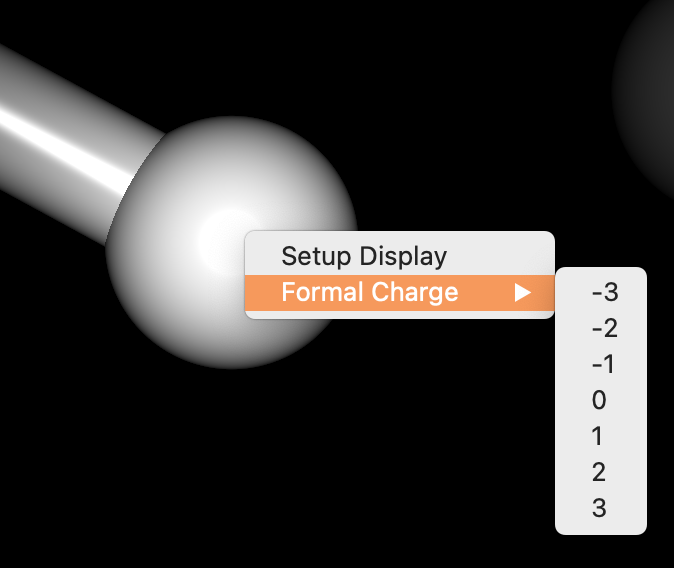
Change of formal charge and bond order
By right-clicking on an atom with the mouse, a pop-up menu appears, allowing you to change the formal charge of that atom.
Move the cursor over the “Formal Charge” menu item that appears to display the options for selecting a charge. Move the cursor over the desired formal charge from the options displayed and then release the mouse button. The formal charge of the selected atom will then be changed.
To check, see the “Object Inspector” dialog in Interface.
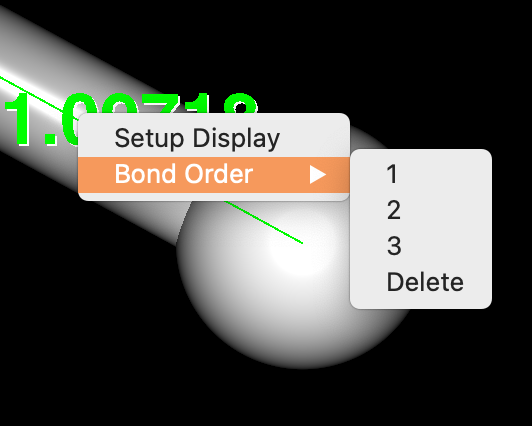
By right-clicking on a bond with the mouse, a pop-up menu appears, allowing you to change the multiplicity of the bond.
Move the cursor over the “Bond Order” menu item that appears, and you will be presented with options for selecting the bond multiplicity. Move the cursor over the desired option and release the mouse button. The multiplicity of the selected bond will then be changed and the molecular display will be updated.
If you choose “Delete” from the options, the bond will be deleted.
When a file with a crystal structure is opened and displayed in packing view, any changes made will apply to all copies of itself that are displayed in packing view, not just the selected atoms and bonds.
Controller
Clicking the [Controller] button in the [Molecule Box] dialog, opens the toolbar for controlling the molecular display. The content of the toolbar changes according to the opened molecule file (see below).
The "Init.", "Axis", "Fitting", and "Superimpose" buttons are always displayed. The function of each button is as follows
- Axis
- On/Off display of X, Y, Z axes in molecular display
- Fitting
- When opening a coformations file, the display of molecules is switched to the coordinates of the entire conformer aligned to minimize RMSd or to the original coordinates.
- Superimpoise
- When opening an conformers file, select multiple atoms and redo the superposition based on them.
- Init.
- Reset rotation, scaling, and translation of the molecule to its initial state
Next to this, "X:", "Y:", and "Z:" boxies are displayed to indicate the rotation status of the molecule.
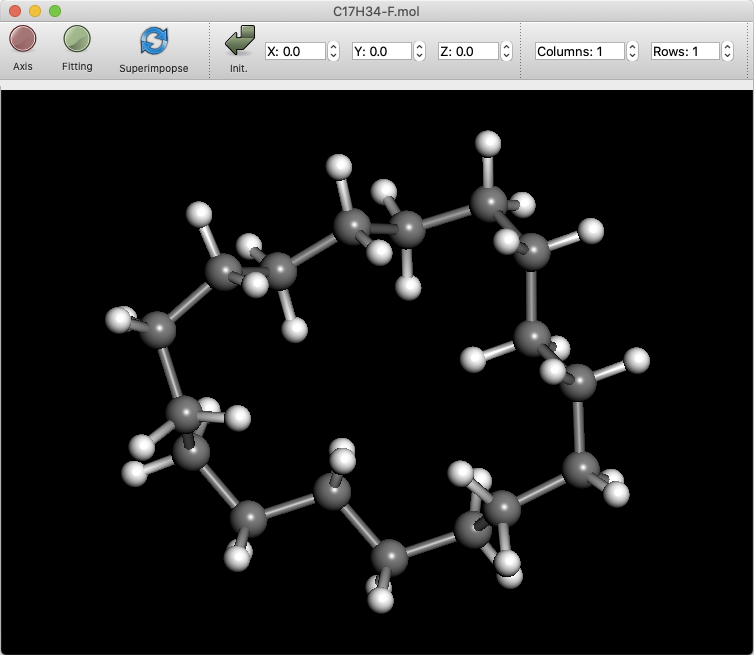
The “Columns” and “Rows” boxes will then appear. These numbers can be increased to split the molecule display. The figure shows a 2X2 display, with the display format of each split window changed.
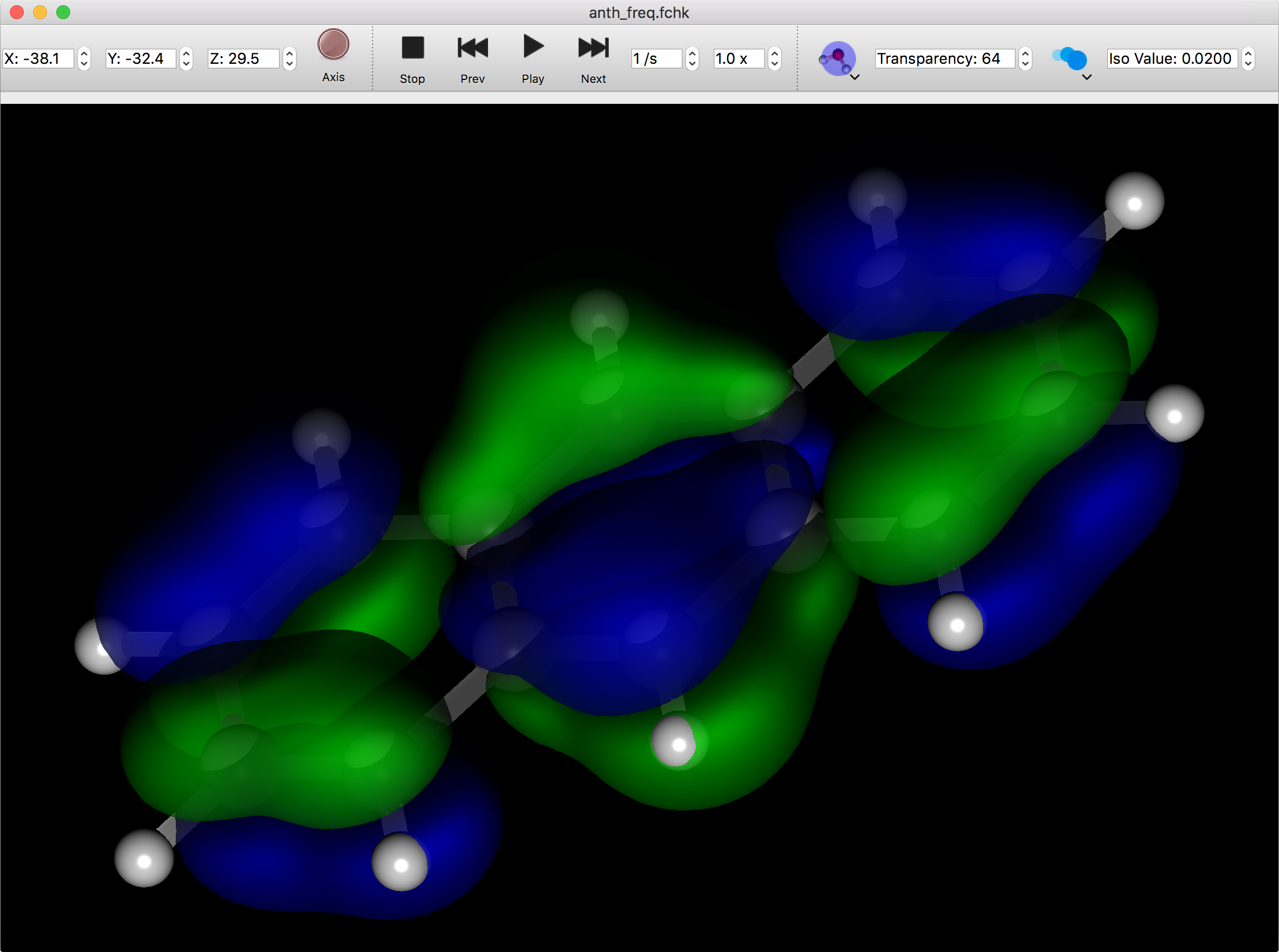
Vibration Animation
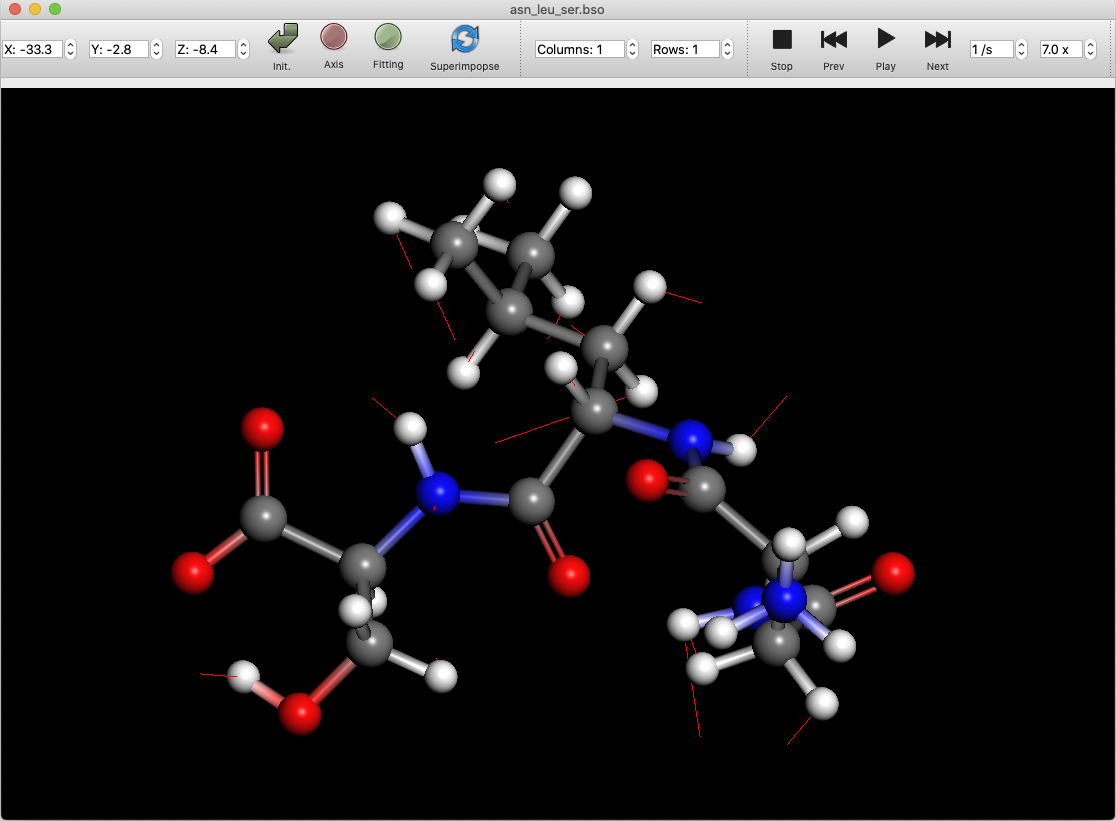
If the molecule file contains vibration information, you can animate the vibration. Click on the “Play” button to start the animation. Click the “Next” or “Prev” buttons to advance or reverse the animation one step at a time.
Click the “Stop” button to return to the initial state before the animation.
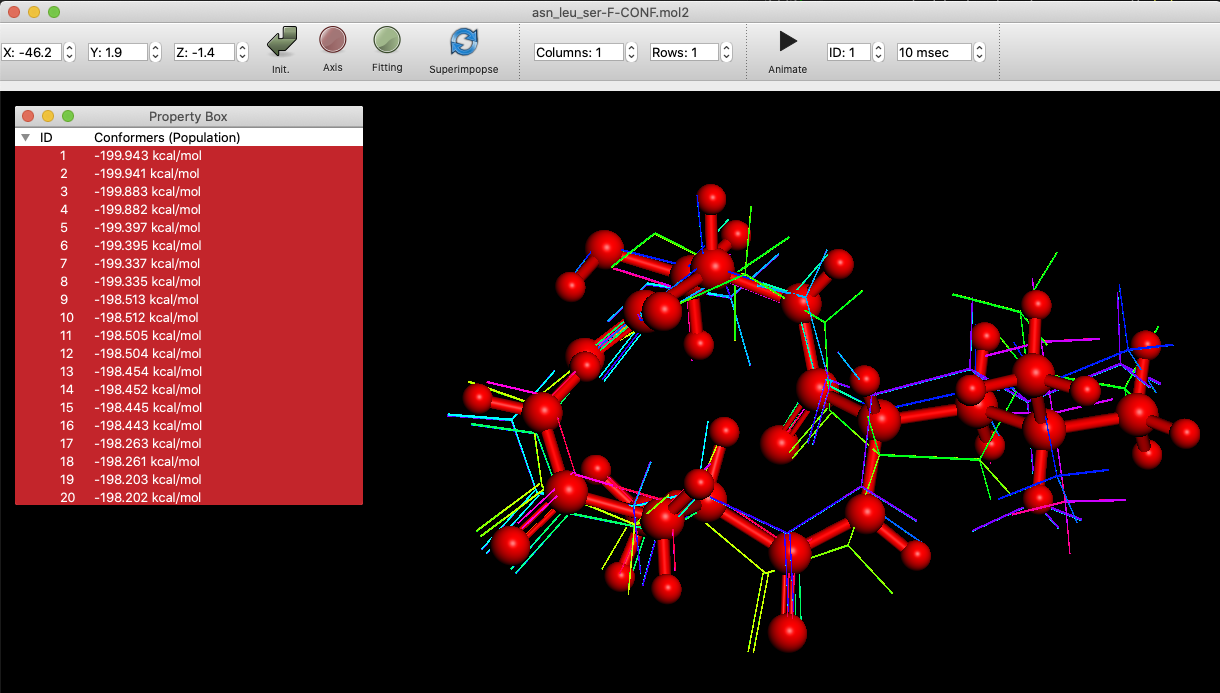
Superimpose
When an SDF file is opened, it is possible to superimpose the conformers selected in the Propety Box.
The conformers are moved and rotated in order to overlap each atom maximally. To display the original data, please click the “Fitting” button, and turn it off.
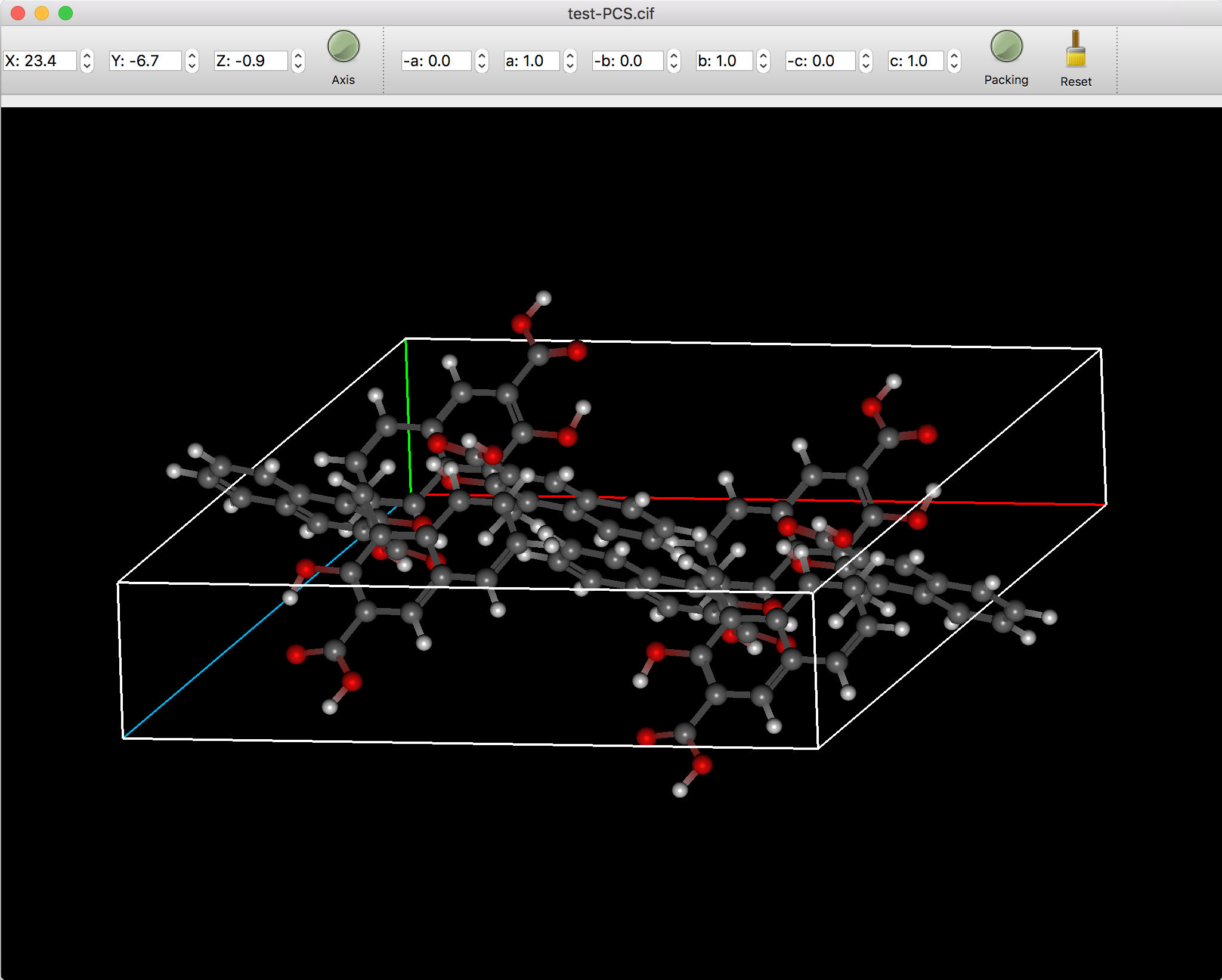
Crystal display
When the crystal file is opened, it is linked with the [Packing] button, and when the [Packing] button is clicked, the packing of the crystal is displayed. “Axis” is also turned on and the cell axes are displayed. In this case, clicking the Axis button will hide the cell axes. The packing display mode allows you to set an additional number of packing repeats in each of the a, b, and c directions to display a larger packing status.
Molecular Orbital
When you open an FChk file containing molecular orbital information, additional buttons appear to set the size of the molecular orbital and the transparency of the surface.