Introduction
Thank you for purchasing CONFLEX.
CONFLEX is a program for the calculation of
- conformation space search
- crystal structure prediction
- normal vibration analysis
- thermodynamic quantities
- ultraviolet and visible light absorption spectra
- circular dichroism spectra
- NMR coupling constants
etc...
CONFLEX algorithms take into account conformational searches and conformational distribution for organic molecules using molecular mechanics. Many users find CONFLEX to be a powerful tool for carrying out various computational studies requiring three-dimensional conformation and analysis of flexible molecules. This manual provides an explanation of how to use CONFLEX.
Main Functions
Conformation Search
CONFLEX automatically recognizes the cyclic and linear parts of the molecule, and creates the initial structure by performing Corner Flap and Edge Flip for the cyclic part and Stepwise Rotation for the linear part. Then, geometry optimization is performed for all of them, and the resulting conformational isomers are stored.
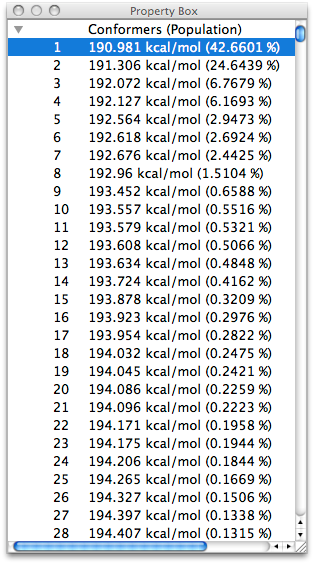
One of the purposes of the conformation search is to calculate the most stable structure of the target molecule, but if the molecular structure is flexible, the number of conformations obtained by the search may be very large. However, if the molecular structure is flexible, the number of possible conformations can be very large. Therefore, CONFLEX uses a reservoir algorithm that always creates the initial structure from energetically stable conformations, and also allows the user to specify the range of energy difference from the most stable structure to be used as a trial structure.
These two features improve the efficiency of the search for the most stable structure and prevent an explosive increase in the number of conformers.
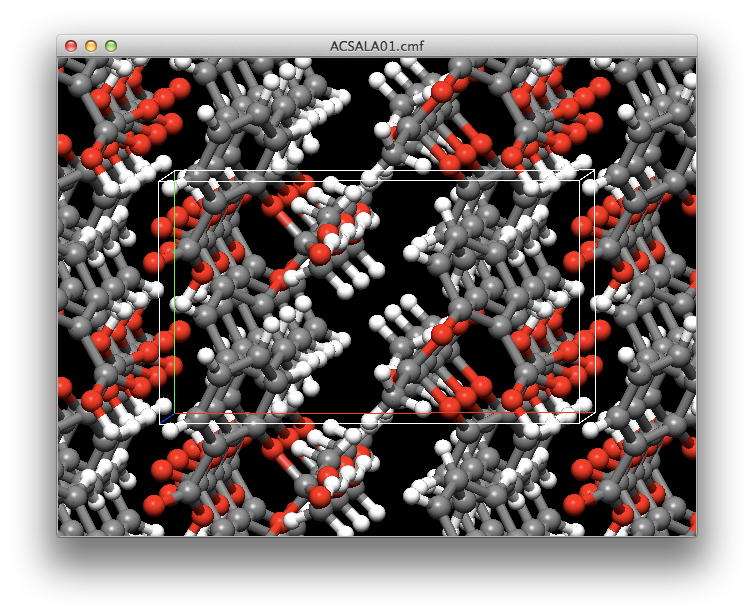
Crystal Structure Prediction
CONFLEX automatically generates the crystal structure using the molecular structure data and the symmetry of the space group. Then, performs structural optimization to comprehensively calculate the crystal structure located at the energy minimum.
In addition to sorting the optimized crystal structures in order of decreasing energy, it is also possible to sort them in order of proximity to previously prepared powder diffraction data.
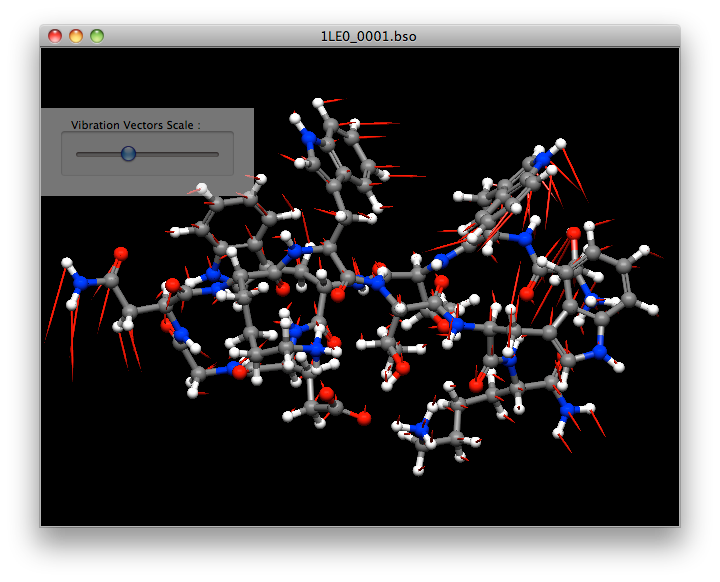
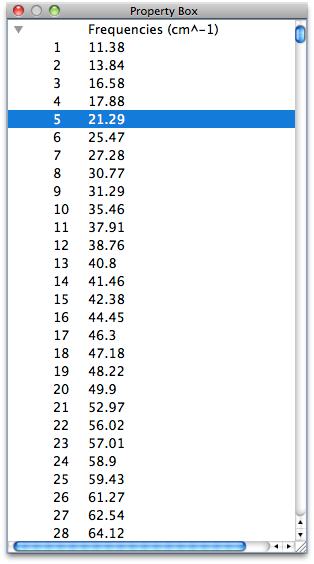
Normal Vibrational Analysis
CONFLEX automatically performs a vibration analysis on the structure obtained by geometry optimization. It also calculates thermodynamic quantities such as Gibbs free energy.
Working with Gaussian
If you have Gaussian installed on the same machine, CONFLEX can call Gaussian to perform geometry optimization and conformation search calculations.
In addition to the above, it is possible to use Gaussian for molecules without molecular force field parameters or for electronic states that cannot be handled by classical force fields.
Dynamic Reaction Coordinate (DRC)
DRC is a calculation method that uses the reference vibration mode to calculate the initial velocity vector and perform dynamics calculations.
It can be applied to the configuration transformation of multiple molecules and the conformation transformation of large molecules.
Host-Ligand coordination search
For systems containing multiple molecules, CONFLEX search for the position and orientation of one molecule in relation to other molecule(s) (groups) to determine its stability.
It can be used to search for stable structures of dimers or complexes.
Solvent Effect
The GB/SA model can be used for structural optimization, vibration analysis, and conformation analysis.
In addition, LogP values can be calculated automatically.
Parameter Configuration
For molecules containing atom types that are not supported by the force field parameters included in CONFLEX, additional parameters can be added to the calculation.
Only the MMFF94s force field is supported.
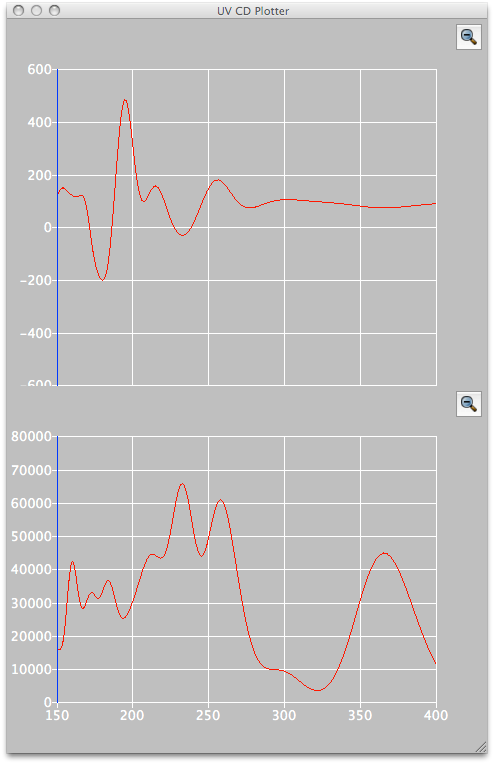
CD/UV/Vis Spectral Analysis
CD/UV/Vis spectral calculations can be performed for the conformational isomers obtained by CONFLEX.