How to execute CONFLEX
[Input files]
A structure file containing molecular or crystal structure data and an ini file containing keywords for calculation settings are used as input files. You can use the structure file of MOL/MOL2/PDB/CIF/CMF format. The names of the structure and ini files have to match, expect for thier file extensions. The ini file have to have extention of “.ini”. These input files have to be stored in a single folder when executing CONFLEX. If you run a CONFLEX job by CONFLEX Interface, the ini file is automatically created by the program. On the other hand, if you execute a CONFLEX job from command line, you need to make the ini file manually in advance. You can also execute a CONFLEX job without the ini file. In that case, calculations of molecular geometry optimization and normal mode analysis are performed with default settings.
Execution from CONFLEX Interface (Windows/Mac/Linux)
Launch CONFLEX Interface.
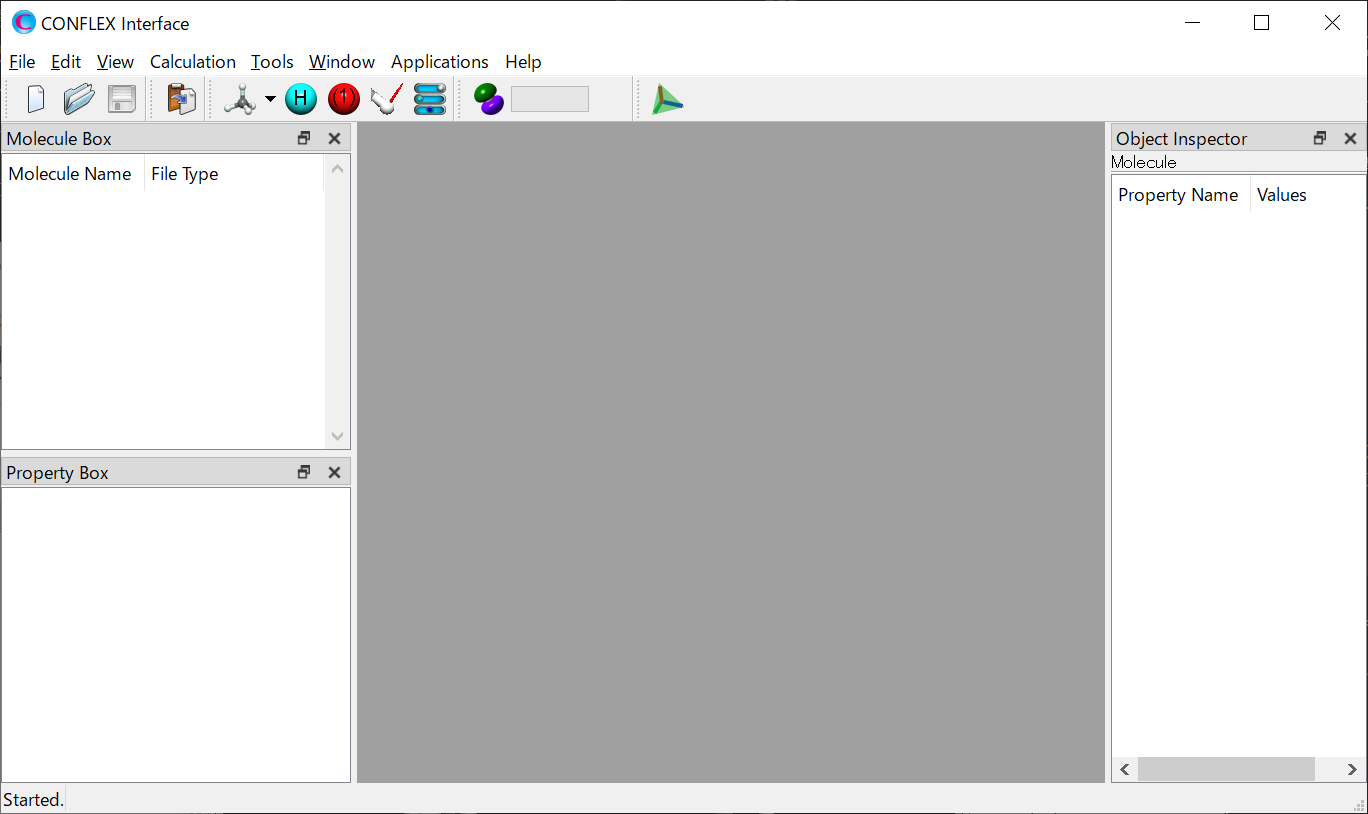
Either select [Open] from the File menu and specify the structure file or drug & drop the structure file into the gray part of the CONFLEX Interface. The molecule will be displayed.
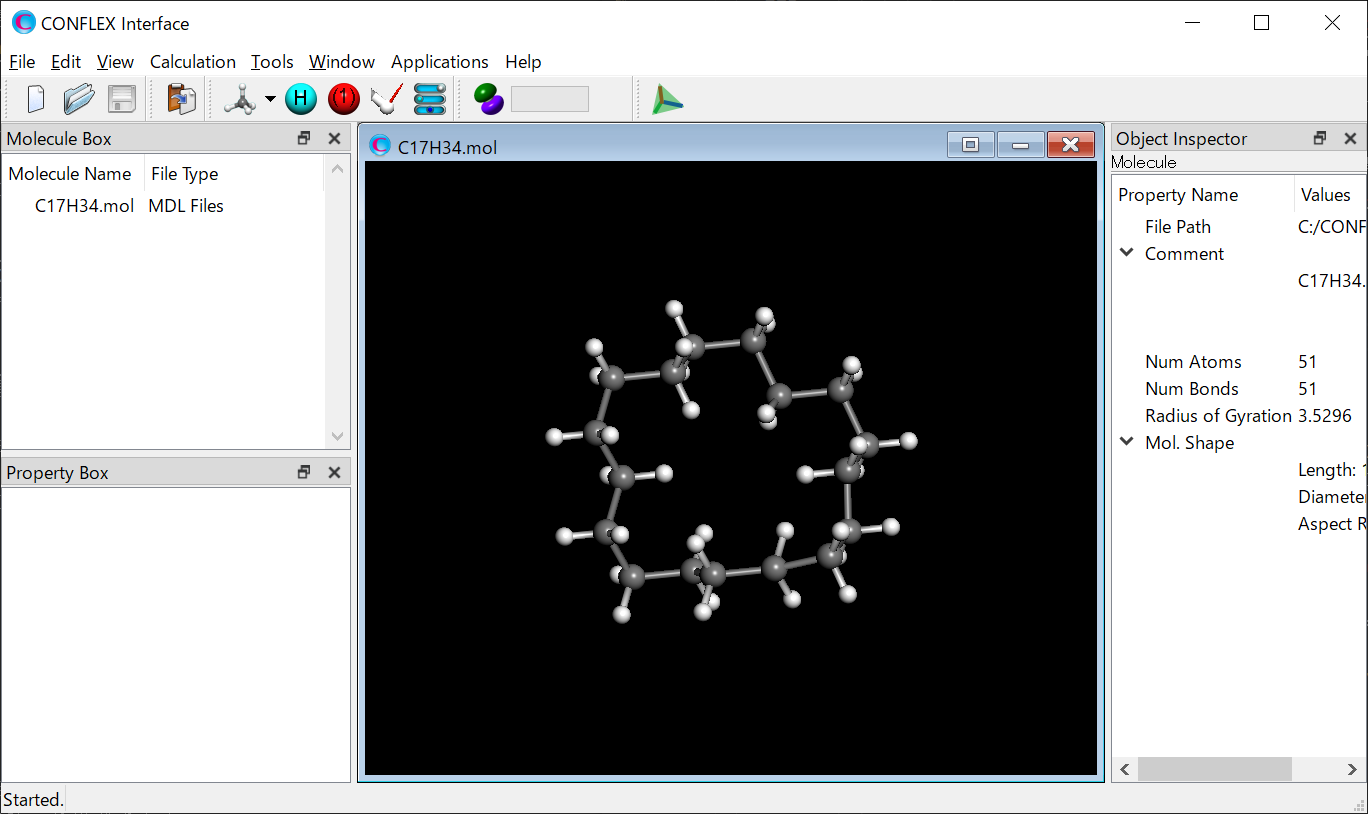
Next, select [CONFLEX] from the Calculation menu. The calculation setting dialog will be displayed.
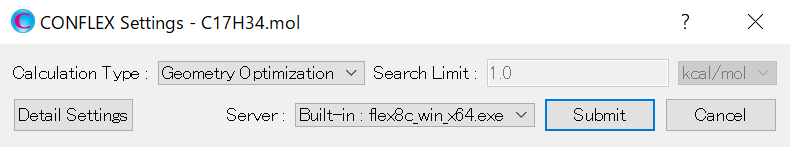
Click . Calculations of molecular geometry optimization and normal mode analysis will be performed in default setting.
If you want to perform other calculations, either select an other option from the pull-down menu of [Calculation Type:] or click to open the detailed settings dialog.
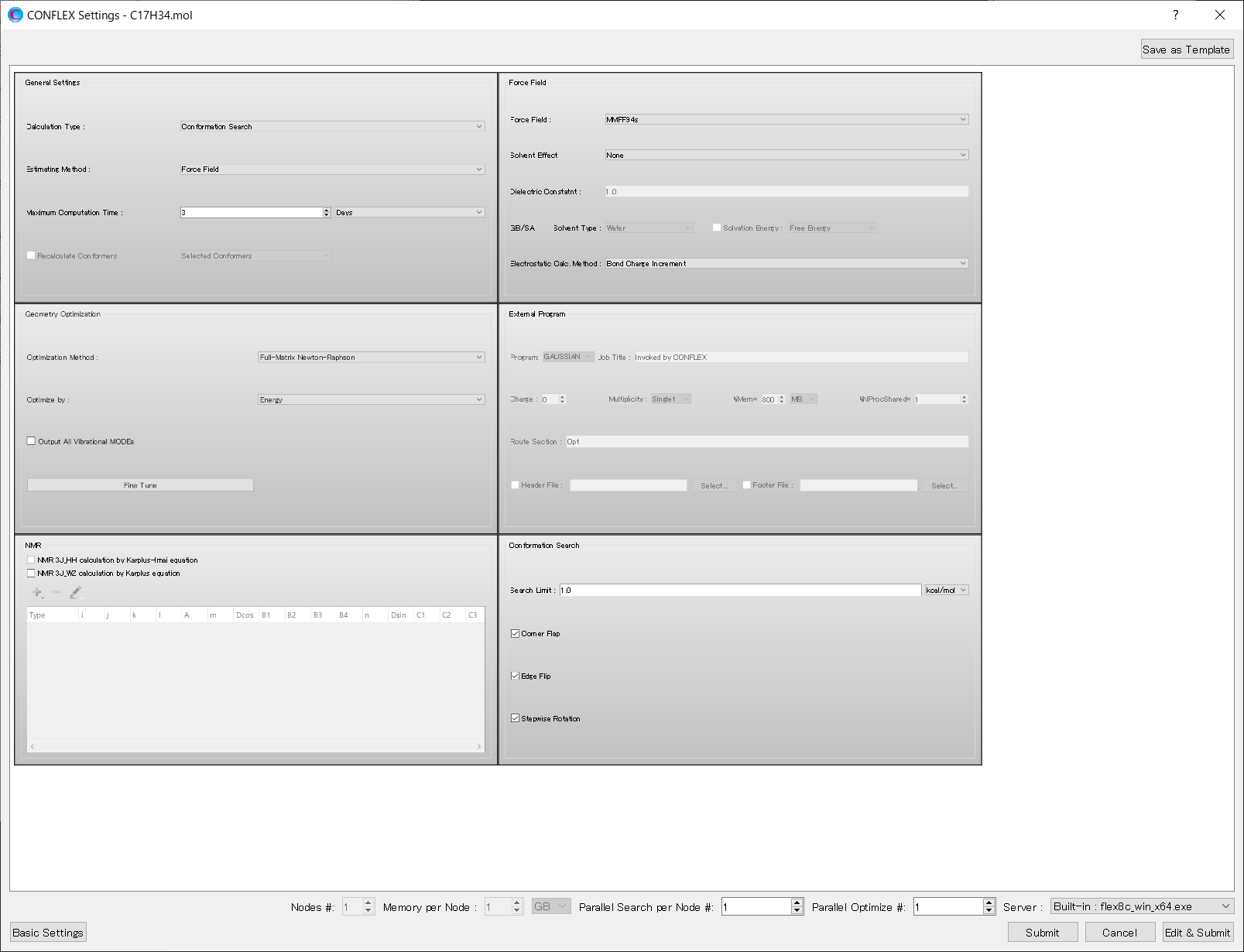
In the [General Settings] dialog in the detailed settings dialog, you can select a different calculation from the pull-down menu of [Calculation Type:].
The detailed settings dialog allows you to change various calculation settings. For more information on using this dialog, please refer to detail setting manual of CONFLEX Interface.
After completing the settings, click to start the calculation.
On the other hand, if you click , the following dialog will open.
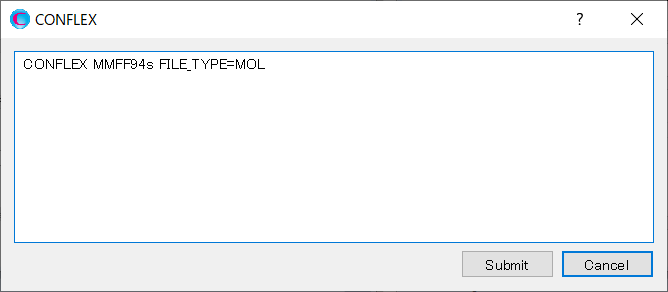
The calculation settings are defined by specifying keywords in the (structure filename).ini file. The CONFLEX Interface automatically generates the (structure filename).ini that describes the contents in the dialog. You can edit the contents directly in the dialog. After completing the editing, click to start the calculation.
[How to use Parallel CONFLEX]
In the detailed settings dialog, you can configure settings of parallel calculation.
- The [Parallel search per Node#:] specifies the number of processes for search calculations parallelized by MPI.
- The [Parallel Optimize #:] specifies the number of threads for optimization calculations parallelized by OpenMP.
In a conformation search and crystal structure search, both parallelizations can be used simultaneously. In this case, the job will use the number of CPU cores equal to the product of [Parallel search per Node#:] and [Parallel Optimize #:] values.
[Execution from command line (Windows)]
Prepare the structure file and the ini file as input files. You can use the structure file of MOL/MOL2/PDB/CIF/CMF format. The names of the structure and ini files have to match, expect for thier file extensions. Here, we use Cyclohexane.mol and Cyclohexane.ini files in [C:\CONFLEX\Sample_Files\CONFLEX\optimization_and_search]. Create a “Test” folder on Desktop, and copy both files into it.
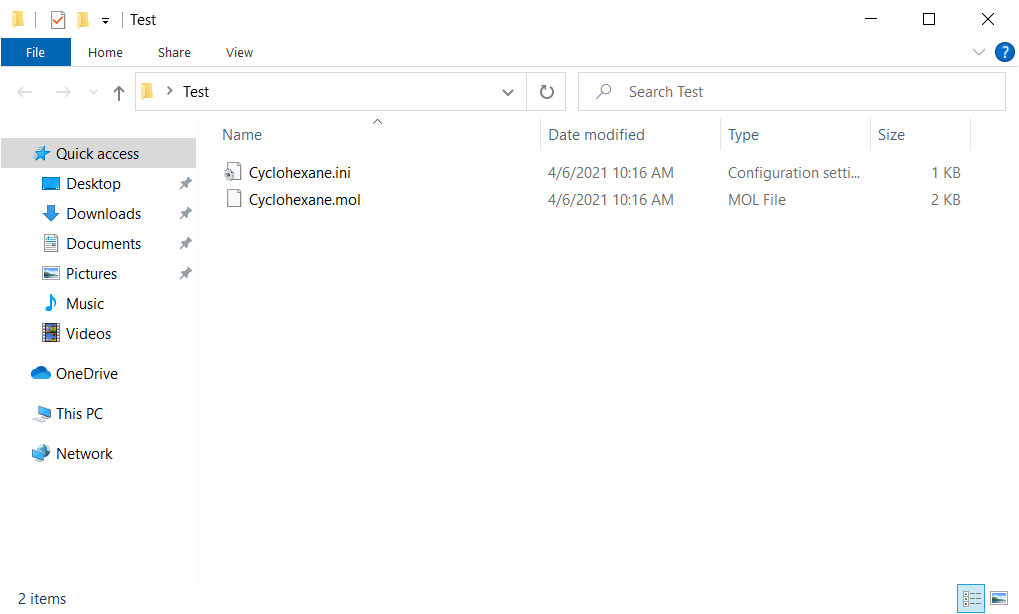
The keywords for calculation settings are described in the Cyclohexane.ini file. About explanations of the keywords, please refer to a manual.
Next, launch a command prompt
Run the following command to change position from current directory to Test folder.
cd C:\Users\conflex\Desktop\Testenter
* The “conflex” in the address above is the account name. For example, if your account name is “guest”, the address would be “C:\Users\guest\Desktop\Test.”
Next, run the following command to list files in the Test folder.
direnter
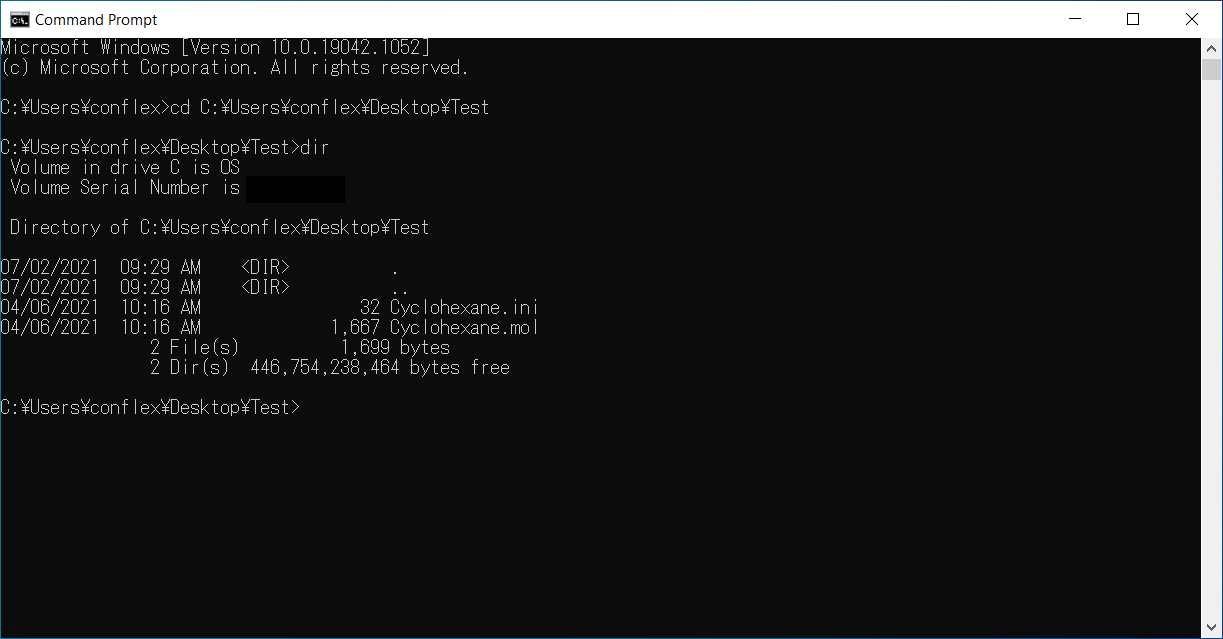
There are two files, Cyclohexane.mol and Cyclohexane.ini.
Run the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par Cyclohexaneenter
* The “-par” option in the command specifies address of folder containing parameter files. The last argument in the command is the name of structure file excluding the extension.
[How to use Parallel CONFLEX]
The number of threads for optimization calculation is set using the “-omp” option in the CONFLEX program. The number of processes for search calculation is set using the “-np” option in the “mpiexec.exe” program. When using the mpiexec.exe command, you should execute the following command first to set up the MPI environment.
C:\CONFLEX\bin\vars.batenter
If you want to parallelize an optimization calculation, run CONFLEX using the following command.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par -omp [the number of threads] [structure file name(without extension)]enter
If you want to parallelize a search calculation, run CONFLEX using the following command. Note that the CONFLEX program name in the following command is different from that in the command above.
C:\CONFLEX\bin\mpiexec.exe -np [the number of processes] C:\CONFLEX\bin\conflex_mpi-10a.exe -par C:\CONFLEX\par [structure file name(without extension)]enter
If you want to parallelize both the search and optimization calculations, run CONFLEX with the following command.
C:\CONFLEX\bin\mpiexec.exe -np [the number of processes] C:\CONFLEX\bin\conflex_mpi-10a.exe -par C:\CONFLEX\par -omp [the number of threads] [structure file name(without extension)]enter
Each parallelization of optimization and search is implemented using OpenMP and MPI, respectively. The “-omp” and “-np” options are used to set the number of parallels in OpenMP and MPI, respectively.
[Execution from command line (Linux)]
Prepare the structure file and the ini file as input files. You can use the structure file of MOL/MOL2/PDB/CIF/CMF format. The names of the structure and ini files have to match, expect for thier file extensions. Here, we use Cyclohexane.mol and Cyclohexane.ini files in [/usr/local/conflex/Sample_Files/CONFLEX/optimization_and_search/].
First, launch a terminal.
Run the following commands to create a “Test” folder in Home directory and copy both files into the Test folder.
mkdir ~/Testentercp /usr/local/conflex/Sample_Files/CONFLEX/optimization_and_search/Cyclohexane.mol ~/Testentercp /usr/local/conflex/Sample_Files/CONFLEX/optimization_and_search/Cyclohexane.ini ~/Testenter
Run the following command to change position from current directory to the Test folder.
cd ~/Testenter
Next, execute the following command to list the files in the Test folder.
ls -lenter
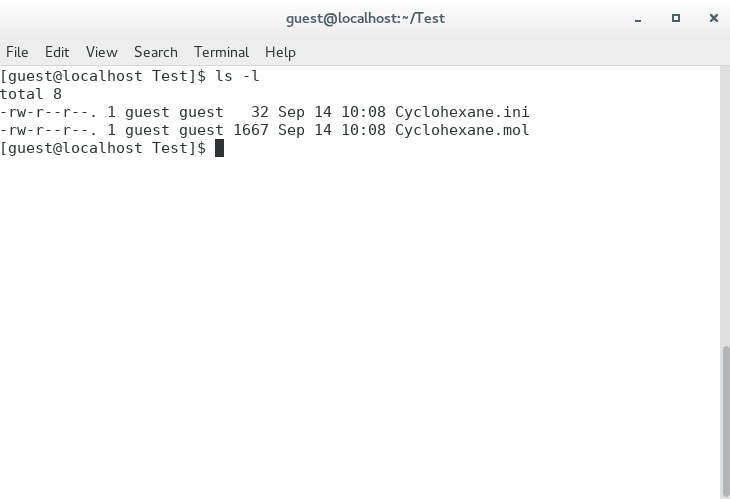
There are two files, Cyclohexane.mol and Cyclohexane.ini. The keywords for calculation settings are described in the Cyclohexane.ini file. About explanations of the keywords, please refer to a manual.
Run the following command to start the calculation.
/usr/local/conflex/conflex-10a.exe -par /usr/local/conflex/par Cyclohexaneenter
* The “-par” option in the command specifies address of folder containing parameter files. The last argument in the command is the name of structure file excluding the extension.
[How to use Parallel CONFLEX]
The number of threads for optimization calculation is set using the “-omp” option in the CONFLEX program. The number of processes for search calculation is set using the “-np” option in the “mpiexec.exe” program.
If you want to parallelize an optimization calculation, run CONFLEX using the following command.
/usr/local/conflex/conflex-10a.exe -par /usr/local/conflex/par -omp [the number of threads] [structure file name(without extension)]enter
If you want to parallelize a search calculation, run CONFLEX using the following command. Note that the CONFLEX program name in the following command is different from that in the command above.
/usr/local/conflex/bin/mpirun -np [the number of processes] /usr/local/conflex/bin/conflex_mpi-10a.exe -par /usr/local/conflex/par [structure file name(without extension)]enter
If you want to parallelize both the search and optimization calculations, run CONFLEX with the following command.
/usr/local/conflex/bin/mpirun -np [the number of processes] /usr/local/conflex/bin/conflex_mpi-10a.exe -par /usr/local/conflex/par -omp [the number of threads] [structure file name(without extension)]enter
Each parallelization of optimization and search is implemented using OpenMP and MPI, respectively. The “-omp” and “-np” options are used to set the number of parallels in OpenMP and MPI, respectively.
[Execution from command line (Mac)]
Prepare the structure file and the ini file as input files. You can use the structure file of MOL/MOL2/PDB/CIF/CMF format. The names of the structure and ini files have to match, expect for thier file extensions. Here, we use Cyclohexane.mol and Cyclohexane.ini files in Sample_Files folder.
First, launch a terminal. The program is in [Application] → [Utility] → [Terminal.app].
Run the following commands to create a “Test” folder in Home directory and copy both files into the Test folder.
mkdir ~/Testentercp /Applications/CONFLEX/Sample_Files/CONFLEX/optimization_and_search/Cyclohexane.mol ~/Testentercp /Applications/CONFLEX/Sample_Files/CONFLEX/optimization_and_search/Cyclohexane.ini ~/Testenter
Run the following command to change position from current directory to the Test folder.
cd ~/Testenter
Next, execute the following command. The files in the Test folder will be listed.
ls -lenter
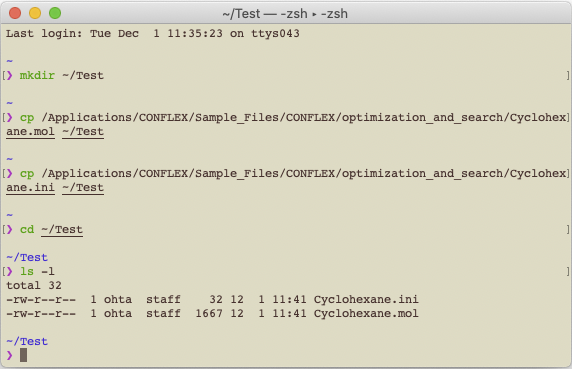
There are two files, Cyclohexane.mol and Cyclohexane.ini. The keywords for calculation settings are described in the Cyclohexane.ini file. About explanations of the keywords, please refer to a manual.
Run the following command to start the calculation.
/Applications/CONFLEX/bin/conflex-10a.exe -par /Applications/CONFLEX/par Cyclohexaneenter
* The “-par” option in the command specifies address of folder containing parameter files. The last argument in the command is the name of structure file excluding the extension.
[How to use Parallel CONFLEX]
The number of threads for optimization calculation is set using the “-omp” option in the CONFLEX program. The number of processes for search calculation is set using the “-np” option in the “mpiexec.exe” program.
If you want to parallelize an optimization calculation, run CONFLEX using the following command.
/Applications/CONFLEX/bin/conflex-10a.exe -par /Applications/CONFLEX/par -omp [the number of threads] [structure file name(without extension)]enter
If you want to parallelize a search calculation, run CONFLEX using the following command. Note that the CONFLEX program name in the following command is different from that in the command above.
/Applications/CONFLEX/bin/mpirun -np [the number of processes] /Applications/CONFLEX/bin/conflex_mpi-10a.exe -par /Applications/CONFLEX/par [structure file name(without extension)]enter
If you want to parallelize both the search and optimization calculations, run CONFLEX with the following command.
/Applications/CONFLEX/bin/mpirun -np [the number of processes] /Applications/CONFLEX/bin/conflex_mpi-10a.exe -par /Applications/CONFLEX/par -omp [the number of threads] [structure file name(without extension)]enter
Each parallelization of optimization and search is implemented using OpenMP and MPI, respectively. The “-omp” and “-np” options are used to set the number of parallels in OpenMP and MPI, respectively.