cifファイルを用いた結晶構造最適化
ここでは、CONFLEXのインストール場所の
CONFLEX/Sample_Files/CONFLEX/crystal/optimization/cif_file
フォルダ内の「tartronicacid.cif」ファイルを例として説明します。
このファイルをご自身のホームディレクトリー以下の適当な場所にコピーして作業を行なってください。
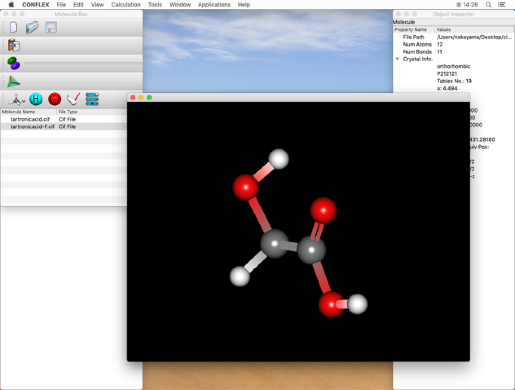
- 「File」メニューから「Open」をクリックし、コピーした「tartronicacid.cif」ファイルを開きます。
-
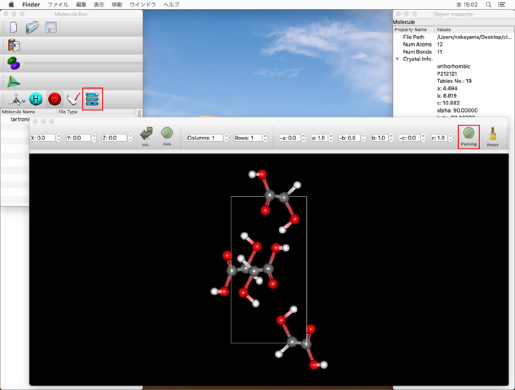
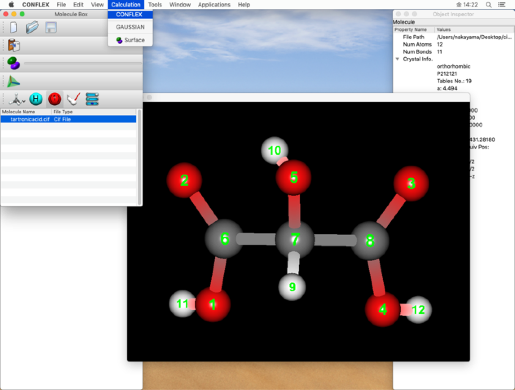
「Molecule Box」ダイアログのLabelアイコン
 をクリックし、原子の通し番号を確認します。
をクリックし、原子の通し番号を確認します。
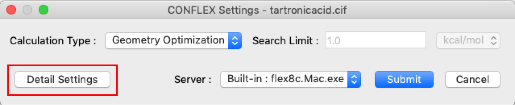
- 「Calculation」メニューから「CONFLEX」を選択し、 CONFLEX Settingsダイアログを表示させます。
- CONFLEX Settingsダイアログの「Detail Settings」をクリックし、詳細設定ダイアログを表示させます。
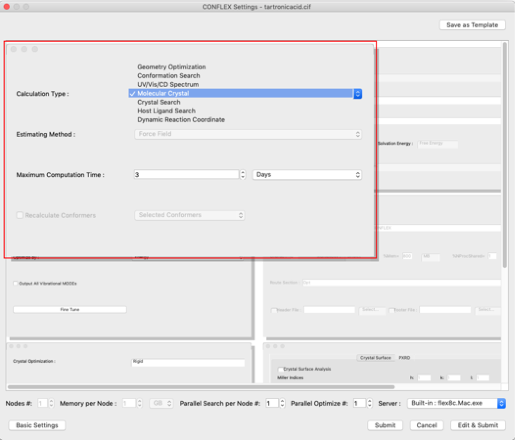
- まず、計算の種類を設定します。
- 次に、最適化方法を設定します。
- 詳細設定ダイアログの「Edit & Submit」をクリックします。
- 分子の結合次数を「CIF_BOND=」キーワードにより設定します。
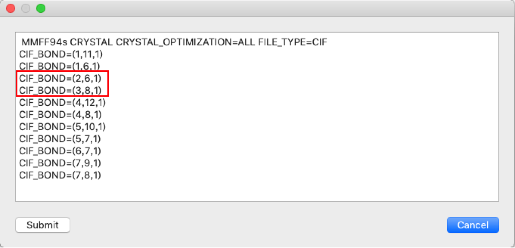
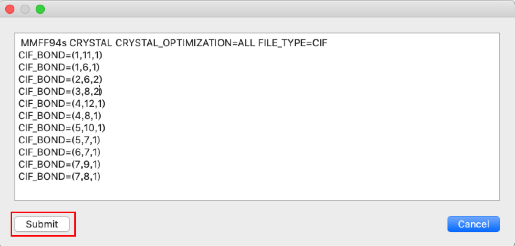
- 結合次数が正しく設定されていることを確認し、「Submit」をクリックします。
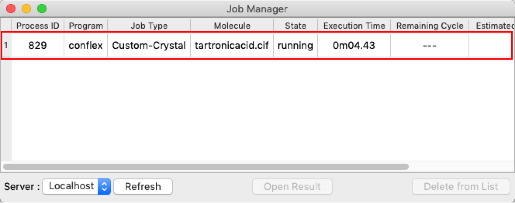
- ジョブが実行されると「Job Manager」が表示されます。
-
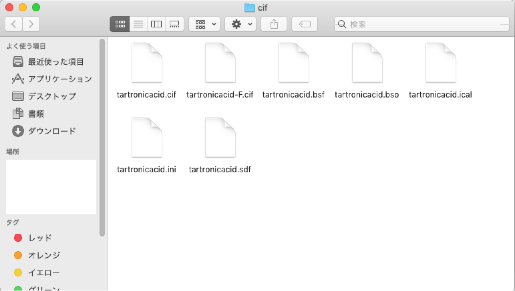
フォルダーには、「tartronicacid.cif」の他に6つのファイル
- tartronicacid.bsf
- tartronicacid.bso
- tartronicacid.ical
- tartronicacid.ini
- tartronicacid.sdf
- tartronicacid-F.cif
が含まれています。
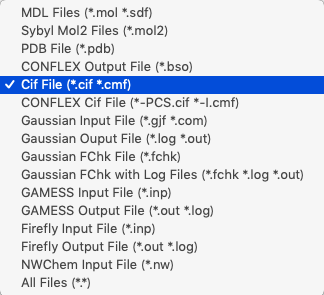
ファイルを開く際、ファイルタイプは「Cif File (*.cif *.cmf)」を選択してください。
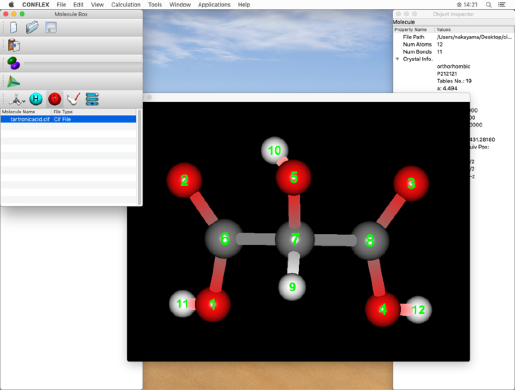
下のステップで二重結合の結合次数を設定しますので、ここで原子番号を確認してください。
左上にある「General Settings」ダイアログの「Calculation Type:」プルダウンメニューから「Molecular Crystal」を選択します。
左下の「Crystal Optimization」ダイアログの「Crystal Optimization:」プルダウンメニューから「ALL」を選択します。格子定数や空間群は、cifファイル内の値が表示されます。
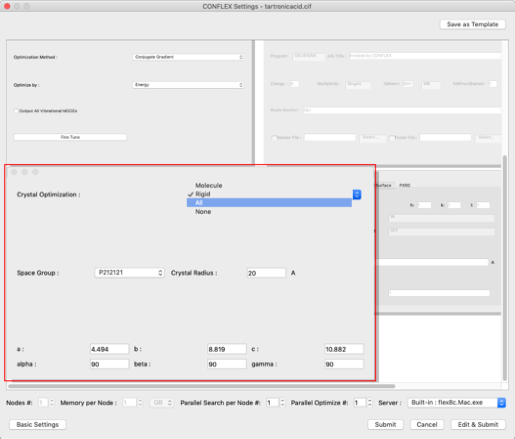
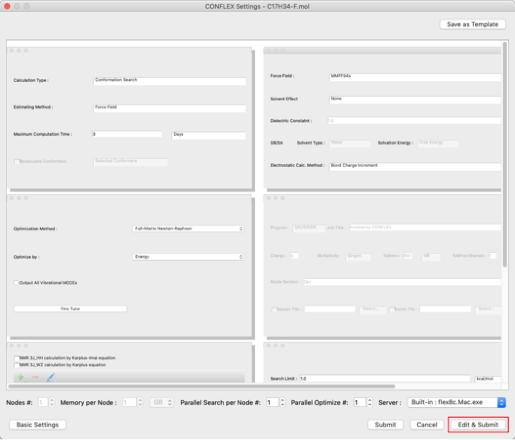
入力分子の2-6、3-8原子間の結合は2重結合ですので、
CIF_BOND=(2,6,1)
CIF_BOND=(3,8,1)
の「1」を「2」に変更します。
* 原子の通し番号は、分子ウィンドウで確認します。
結晶構造最適化ジョブが実行されます。
「State」が「Finished」(ジョブ終了)となっていることを確認したら、赤枠部分をダブルクリックします。
ダブルクリックによって分子ウィンドウが開いて、最適化後の構造(tartronicacid-F.cif)が表示されます。
パッキング構造を表示する場合、Molecule BoxダイアログのControllerアイコン をクリックし、分子ウィンドウに表示されたツールバーにある「Packing」ボタンをクリックします
をクリックし、分子ウィンドウに表示されたツールバーにある「Packing」ボタンをクリックします