はじめに
この度は、CONFLEX DOCKをご購入いただきまして、誠にありがとうございます。
CONFLEX_DOCKは、基質となるタンパク質に対して、指定したペプチド鎖がどこに配位し複合体を形成するかを予測するドッキングシミュレーションプログラムです。
予測する際には、タンパク質のアミノ酸残基を代表点(Cα原子)で粗視化し、ドロネー分割により四面体を構築します。そして、タンパク質表面に設定した探索点にペプチドの残基を置き、四体粗視化ポテンシャルを元にスコアを評価します。
【文献引用】
CONFLEX DOCKで得られた計算結果を論文等に掲載する場合は、以下の文献を引用してください。
T. Yamamoto, Y. Ikabata, H. Goto, “Reconstruction of Four-Body Statistical Pseudopotential for Protein-Peptide Docking”, J. Comput. Chem., Jpn.-Int. Ed., 2024, 10, 2023-0039.
実行方法と入出力ファイル
実行ファイル等の場所
CONFLEX DOCKの実行ファイルとライセンス・パラメーターファイルは以下のフォルダにインストールされます。
| OS | 実行ファイル | ライセンス・パラメーター |
|---|---|---|
| Windows | C:\CONFLEX\bin | C:\CONFLEX\par |
| macOS | /Applications/CONFLEX/bin | /Applications/CONFLEX/par |
| Linux | /usr/local/conflex/bin | /usr/local/conflex/par |
【入力ファイル】
CONFLEX DOCKでは、基質とするタンパク質の構造データが記述されたPDBファイルと、ペプチドの情報が計算時に必要となります。 ペプチド情報は、その構造のデータが記述されたPDBファイルを指定するか、アミノ酸配列の情報を1文字表記、あるいは3文字表記で指定します。 計算設定は拡張子「.ini」としたiniファイルを作成し、各種キーワードを記述することで行います。 CONFLEX InterfaceからCONFLEX DOCKを実行する場合、iniファイルは自動に作成されますが、コマンドラインから実行する場合はあらかじめ自身で作成する必要があります。 iniファイルがなくとも計算は実行できますが、その場合、計算はデフォルト設定にて行われます。
CONFLEX Interfaceからの実行方法
CONFLEX Interfaceを起動します。
次に、基質とするタンパク質のPDBファイルを開きます。
Fileメニューから「Open」を選択し、PDBファイルを指定するか、PDBファイルをCONFLEX Interfaceの灰色部分にドラッグ&ドロップしてください。下図のように、タンパク質が描画されます。
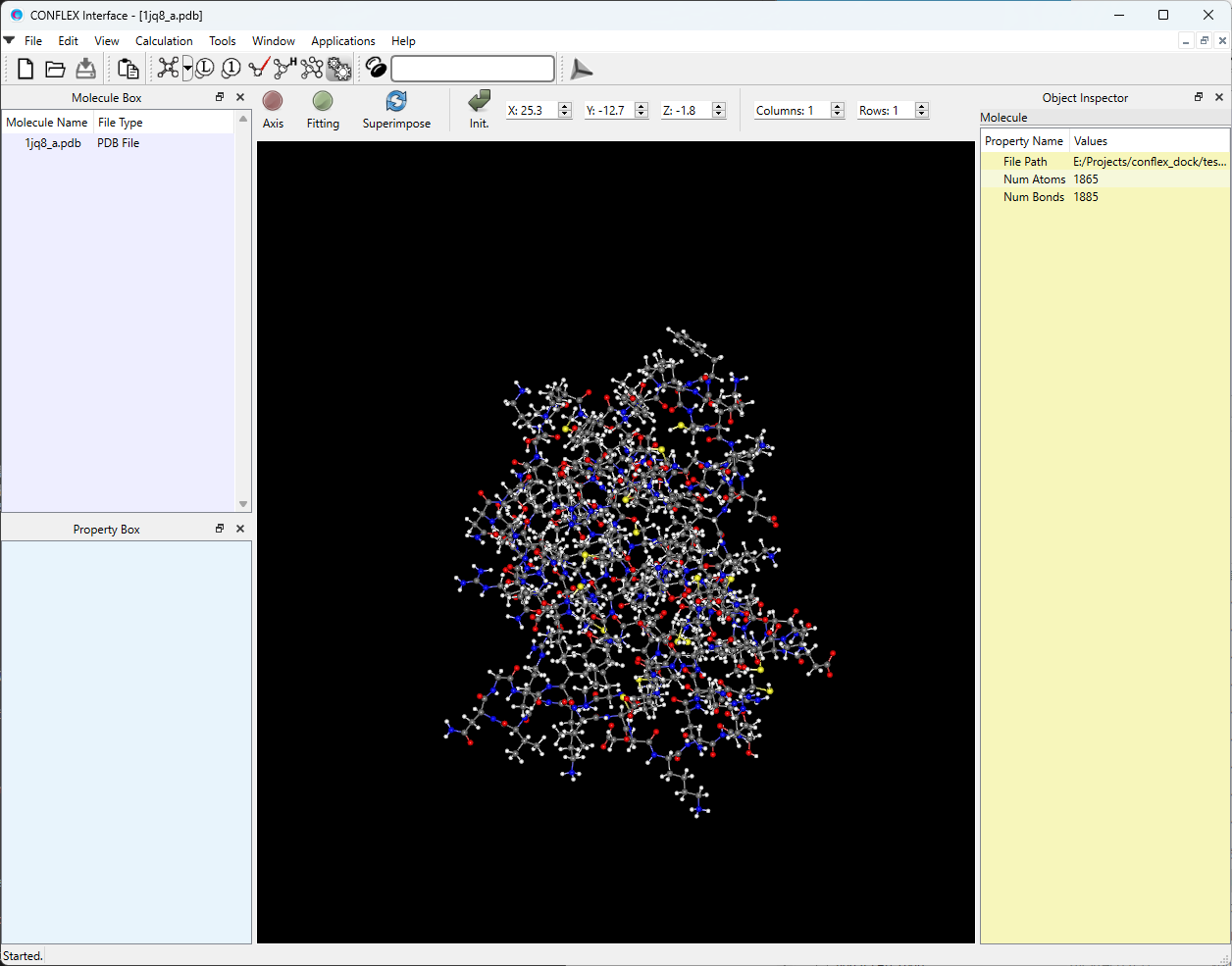
次に、Calculationメニューから「Docking」を選択します。計算設定ダイアログが開きます。
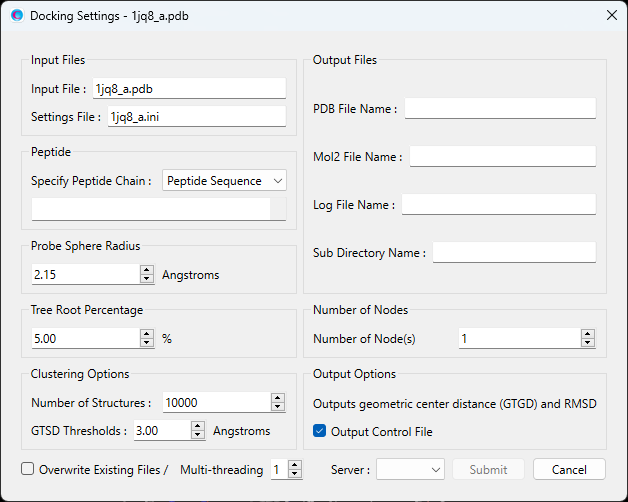
ペプチドの情報は、「Peptide」の項目で設定します。
プルダウンメニューから「Peptide Sequence」を選択し、1文字表記、あるいは3文字表記にて、アミノ酸配列を白枠に記述してください。
3文字表記を用いる場合は、各アミノ酸残基をハイフン“-”で区切ってください。
また、プルダウンメニューから「Open File」を選択することで、ペプチドの情報をPDBファイルで指定することができます。
「Open File」ボタンをクリックして、該当するPDBファイルを指定してください。
その他、本ダイアログでは、各種計算設定や出力ファイル名の指定なども可能です。
計算設定が終わりましたら、をクリックしてください。計算が始まります。
コマンドによる実行方法
コマンドで計算を実行する場合は、“-par”オプションによりライセンス・パラメーターファイルが保存されたフォルダーを指定し、また、“-ipro”オプションによりタンパク質のPDBファイルを、“-ipep”オプションによりペプチドファイル名またはアミノ酸配列を指定して実行します。
【Windowsの場合】
C:\CONFLEX\bin\conflex_dock-1a.exe -par C:\CONFLEX¥par -ipro タンパク質ファイル名 -ipep ペプチドファイル名またはアミノ酸配列enter
【macOSの場合】
/Applications/CONFLEX/bin/conflex_dock-1a.exe -par /Applications/CONFLEX/par -ipro タンパク質ファイル名 -ipep ペプチドファイル名またはアミノ酸配列enter
【Linuxの場合】
/usr/local/conflex/bin/conflex_dock-1a.exe -par /usr/local/conflex/par -ipro タンパク質ファイル名 -ipep ペプチドファイル名またはアミノ酸配列enter
“-ipro”と“-ipep”の指定は必須です。他に出力ファイル名や利用する設定ファイルもオプションにより指定することができます。利用できるオプションは以下の通りです。
| コマンドライン引数 | 説明 |
|---|---|
| -par |
ライセンスファイルおよびスコアファイルが入っているディレクトリーを指定する。 指定が無い場合は、カレントディレクトリーが設定される。 |
|
-omp 数値 *Parallel版のみ利用可 |
並列計算のスレッド数を指定する。 |
| -ipro ファイル名 |
タンパク質のPDBファイルを指定する。 指定が無い場合は、計算は停止する。 |
|
-ipep ファイル名 -ipep ペプチド配列 |
ペプチドのPDBファイルか、アミノ酸配列(1文字または3文字表記)を指定する。 指定が無い場合は、計算は停止する。 |
| -ini ファイル名 |
計算設定ファイルを指定する。 指定が無い場合は、全ての計算条件がデフォルトで実行される。 |
| -olog ファイル名 |
logファイル名を設定する。 指定が無い場合は、-iproで指定したファイル名と、-ipepで指定したファイル名またはアミノ酸配列を組み合わせたファイル名が設定される。 |
| -opdb ファイル名 |
ドッキング結果(ポーズ別)を出力するPDBファイル名を設定する。 指定が無い場合は、-iproで指定したファイル名と、-ipepで指定したファイル名またはアミノ酸配列を組み合わせたファイル名が設定される。 |
| -omol2 ファイル名 |
ドッキング結果(ポーズ別)を出力するmol2ファイル名を設定する。 指定が無い場合は、-iproで指定したファイル名と、-ipepで指定したファイル名またはアミノ酸配列を組み合わせたファイル名が設定される。 |
| -odir ディレクトリー名 |
csvファイル等を出力するディレクトリー名を設定する。 指定が無い場合は、-iproで指定したファイル名と、-ipepで指定したファイル名またはアミノ酸配列を組み合わせたディレクトリー名が設定される。 |
| -h/--help | ヘルプを表示する。 |
出力ファイル
タンパク質のファイルをprotein.pdb、ペプチドのファイルをpeptide.pdbとして
/Applications/CONFLEX/bin/conflex_dock-1a.exe -par /Applications/CONFLEX/par -ipro protein.pdb -ipep peptide.pdbと実行した場合、以下のようなファイル名とフォルダー名で計算結果が出力されます。
| ファイル/ディレクトリー名 | 説明 |
|---|---|
| protein_peptide.log | 計算に用いた設定値や、各ドッキングポーズのスコア値、分布、クラスタリング結果等を出力したファイルです。 |
| protein_peptide.pdb | 各ドッキングポーズをPDB形式で出力したファイルです。 |
| protein_peptide.mol2 | 各ドッキングポーズを粗視化した代表点でMol2形式により出力したファイルです。 |
| protein_peptide/ | 計算結果がcsv形式のファイルで出力されています。 |
なお、ペプチドの情報として、アミノ酸配列を下記のように指定した場合、
/Applications/CONFLEX/bin/conflex_dock-1a.exe -par /Applications/CONFLEX/par -ipro protein.pdb -ipep LAIYS出力ファイル名と作成されるフォルダー名は「protein_LAIYS」となります。出力ファイル名や作成されるフォルダー名はコマンドオプションにより変更できます。