ドッキングシミュレーションの流れ
ここは、CONFLEX DOCKを用いてドッキングシミュレーションを行う流れを説明します。
CONFLEX DOCKは、基質となるタンパク質に対して、指定したペプチド鎖がどこに配位し複合体を形成するかを予測するドッキングシミュレーションプログラムです。
予測する際にはまず、タンパク質のアミノ酸残基を代表点(Cα原子)で粗視化し、ドロネー分割により四面体を構築します。続いて、タンパク質表面に設定した探索点にペプチド残基を置き、四体粗視化ポテンシャルを元にスコアを評価します。
【文献引用】
T. Yamamoto, Y. Ikabata, H. Goto, “Reconstruction of Four-Body Statistical Pseudopotential for Protein-Peptide Docking”, J. Comput. Chem., Jpn.-Int. Ed., 2024, 10, 2023-0039
CONFLEX DOCKでは、計算を実行するプログラムとInterfaceが別々に動作していますので、Interfaceを使用してもしなくても計算を実行することが可能です。まずInterfaceからの計算実行を説明してから、コマンド入力による計算実行の方法を示します。
CONFLEX Interfaceには分子を作成する機能はありませんので、既存のPDB形式のファイルを開いて使用します。
PDBファイルを開く
「File」メニューから「Open」を選ぶと、ダイアログが表示されます。
ダイアログ下部の「Files of type」ポップアップで、開くファイルの形式を「PDB File (*.pdb)」にしてから、目的のPDBファイルを開きます。

目的のファイルを選択してから、「Open」ボタンを押すと、ファイルが開かれグラフィック・ウィンドウに表示されます。
ドッキング計算の開始
計算を開始するには、「Calculation」メニューから「Docking」を選択します。選択すると、以下のダイアログが表示されます:
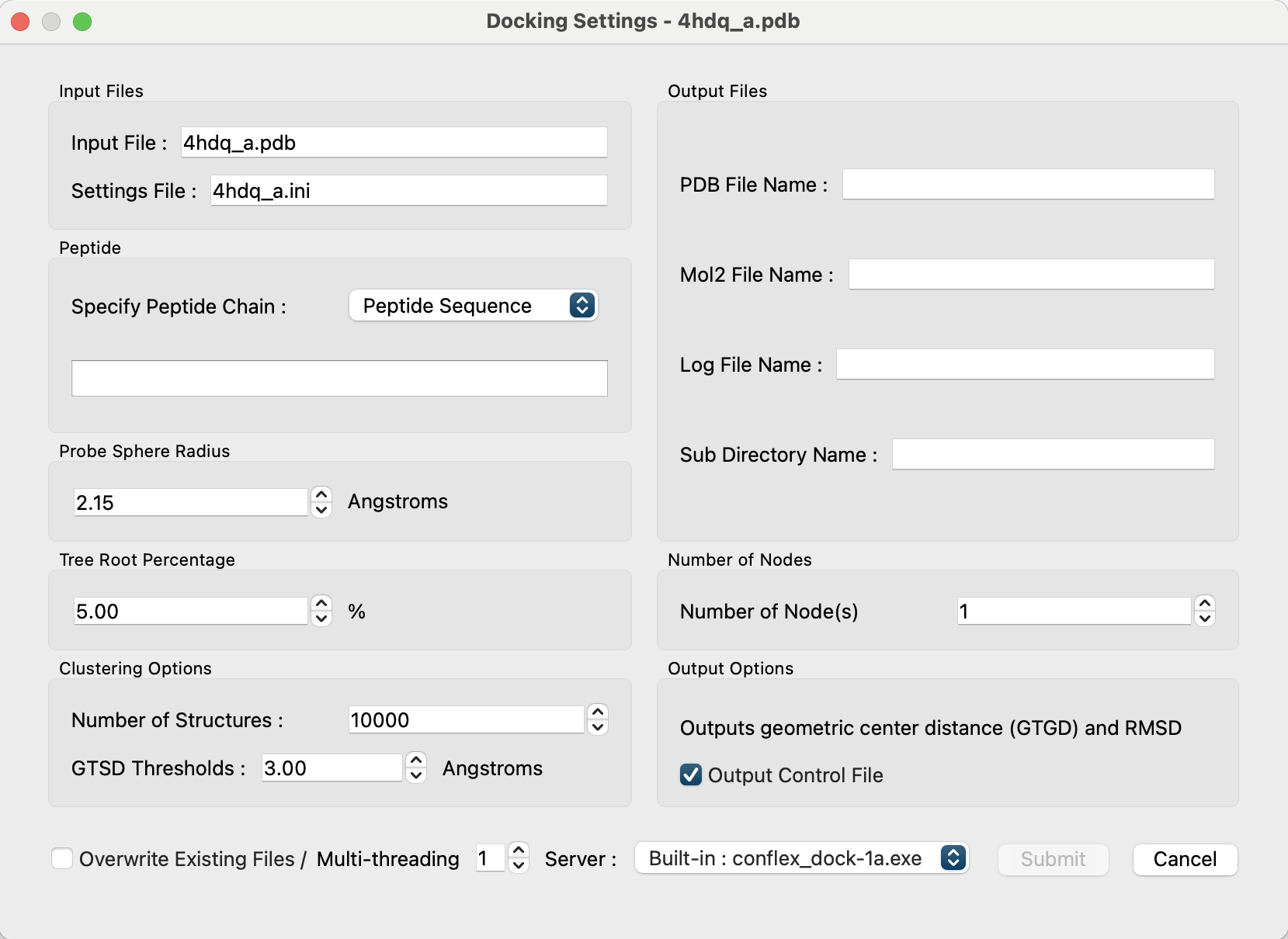
ダイアログの内容を以下で説明します:
- Input Files:
- 計算に使用する入力PDBファイル名および計算設定が保存される入力ファイル名が示されます。
- 「Input File:」には開いていたPDBファイル名が示されます。編集は不可です。
-
「Settings File:」には、このダイアログで設定した内容が保存されます。
デフォルト名として分子ファイル名の拡張子がpdbからiniに変更されたものが示されます。編集して他の名前にすることが可能です。
- Peptide
-
ドッキングを行うペプチドを指定します。
「Specify Peptide Chain:」では、ペプチドの指定方法を選択します。-
「Peptide Sequence」を選択した場合は、すぐ下のボックスにペプチド・シーケンスを入力します。
1文字および3文字標記どちらも認識します。3文字の場合は、アミノ酸の間に「-」を入れてください。
例)ARG-ARG-ASP-TYR-PHE -
「Open File」を選択した場合、「Open File」ボタンが現れます。
クリックするとダイアログが開き、別途用意したペプチドのPDBファイルを開くことができます。
-
「Peptide Sequence」を選択した場合は、すぐ下のボックスにペプチド・シーケンスを入力します。
- Output Files
-
ドッキング計算で出力されるファイルおよびディレクトリー名を指定します。デフォルトでは、開いているタンパク質PDBファイル名本体+「_」+ペプチド鎖表記+拡張子です。
- 「PDB File Name:」ドッキング後の一連の構造ファイルが保存されます。
- 「Mol2 File Name:」ドッキング後の一連の代表点構造が保存されます。
- 「Log File Name:」ドッキング計算のログが保存されます。
- 「Sub Directory Name:」ドッキング計算中に複数のデータファイルが保存されるディレクトリー名です。
- Probe Sphere Radius
- 探索点の設置に使用するプローブ球半径を設定します。
- デフォルトは、2.15Å
- Tree Root Percentage
- 探索点に残基を置きスコアを評価した後,木構造の根として採用し探索を進める点の割合を指定します。
- デフォルトは、5%
- Number of Nodes
- 探索を進めるノードを、スコアの高い順からの個数で選択します。
- デフォルトは1
- Clustering Options
- 「Number of Structures」で、クラスタリングで考慮するスコア上位の予測構造数を指定します(デフォルトは10,000)。
- 「GTSD Thresholds」で、クラスタリングの指標となる、2つの構造のGTGDの閾値を指定します(デフォルトは3.0Å)。
- Output options
- 「Output Control Fileチェックボックス」は、Geometric center To Geometric center Distance (GTGD)とRoot Mean Square Deviation (RMSD)の出力を制御します。
- ペプチド鎖をPDBファイルで指定した場合のみ有効となります(デフォルト:true)
- Overwrite Existing Files
- チェックボックスにチェックを入れると、指定した出力ファイル名が存在しても上書きされます。
- デフォルトはオフ。
- Multi-threading
- OpenMPによる、並列演算数を指定します。
- デフォルトは、1。
- Server:
- 計算を行う実行ファイルを指定します。基本設定は「Preferences...」で行います。
- Submitボタン
- クリックすると計算ジョブへ投入されます。
- 計算の開始は、現在のジョブの状況で判断されます。
Job Manager
計算の「Submit」ボタンをクリックすると、すぐには開始せずジョブ・マネジャーにいったん登録されます。指定したコンピューターのCPUが空いていれば、ジョブが投入されます。 「Job Manager」ダイアログは、計算開始時に自動的に表示されます。 手動で表示させるには、「Tools」メニューの「Job Manager」を選択してください。
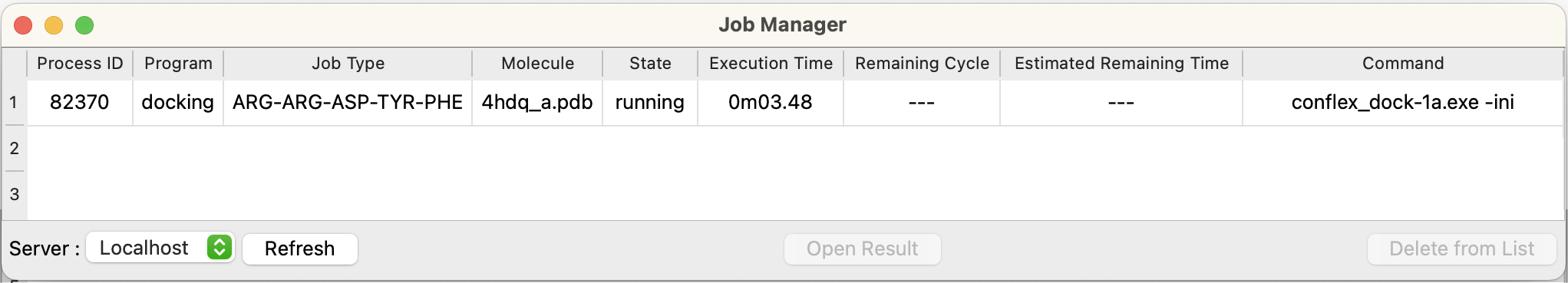
ジョブの経過は、環境設定で指定した秒数ごとにチェックされ、ダイアログの表示が更新されます。自動チェックを指定していない場合は、表示させたいサーバーをダイアログ左下のポップアップから選択し、「Refresh」ボタンをクリックします。「Server:Localhost」が現在Interfaceを起動しているコンピューターを意味します。各項目は、以下の内容になっています。
- Process ID
- 実行されているプログラムの識別番号です。
- Program
- 実行プログラム名が表示されます。
- Job Type
- ドッキングを行なっているペプチドのシーケンスが表示されます。
- Molecule
- 計算している分子のファイル名が表示されます。
- State
- プログラムの実行状況が表示されます。投入時は全て「pending」となります。実行が開始されたら「running」となり、終了したら「Finished」となります。
- Execution Time
- 実行開始してからの経過時間が表示されます。
- Remaining Cycle
- ドッキングの場合は表示されません。
- Estimated Remaining Time
- ドッキングの場合は表示されません。
- Command
- 実行中のコマンドを示します。
計算結果の表示
「Job Manager」内で「Finished」となったジョブは、計算結果を開いて観察する事が可能となります。
「Finished」と表示されているジョブをクリックして選択後、ダイアログ中央下の「Open Result」ボタンをクリックするか、リスト項目をダブルクリックすると計算結果が開きます。
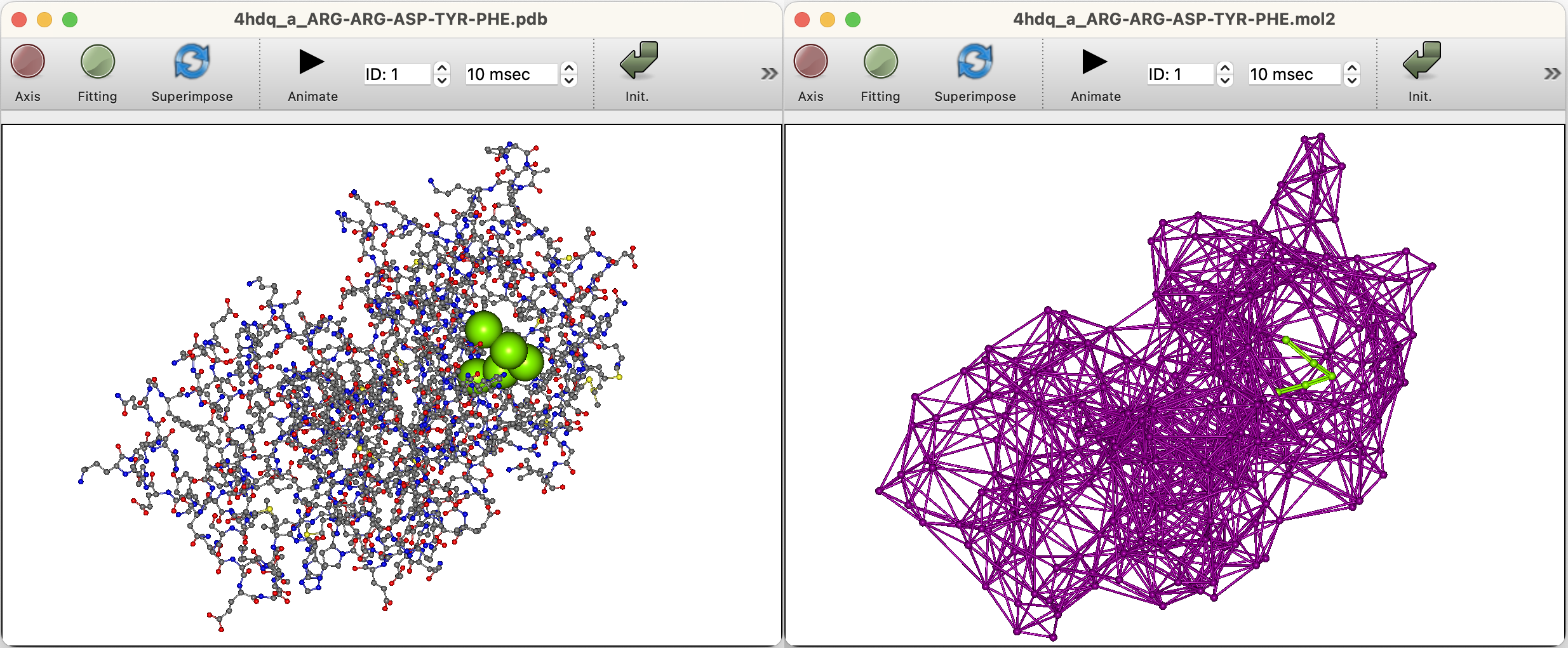
ドッキング計算の場合、計算設定画面で指定したPDB出力ファイルが開きます。
代表点が保存されたMol2ファイルを開きたい場合は、「File」メニューから「Open...」を選択してから、表示されるダイアログでファイルタイプを「Sybyl Mol2 Files (*.mol2)」を指定してから開いてください。
ドッキング・ポーズ・リスト
ドッキング計算結果のファイルを開くと、スコアが最も高い構造がグラフィック・ウィンドウに表示されると同時に、ドッキングスコアの一覧が「Property Box」に表示されます。リストの項目をクリックして選択すると、グラフィック・ウィンドウ内の構造がそのドッキング・ポーズに変更されます。
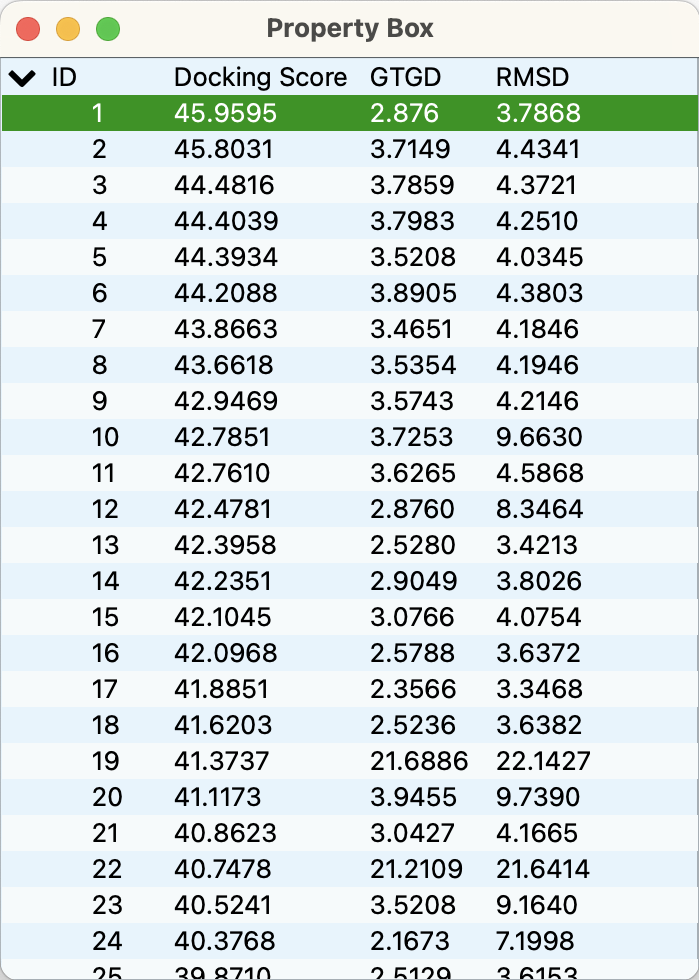
「Property Box」内でシフトキーを押しながらクリックして複数の構造を選択すれば、分子画面で重なった状態で表示させることも可能です。