配座探索
CONFLEXによる配座探索について説明します。
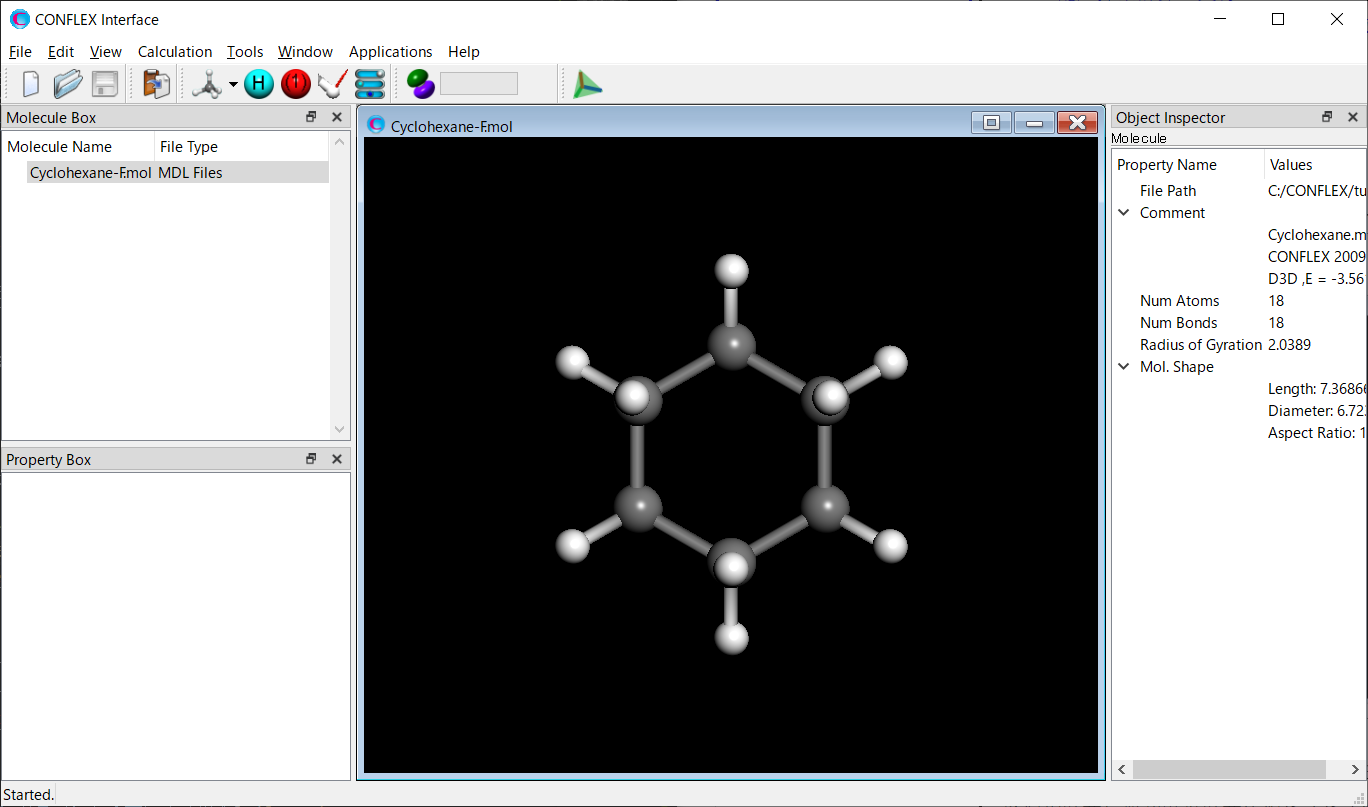
入力構造ファイルは、「構造最適化と振動解析の実行」により得られたCyclohexaneの最適化構造ファイルCyclohexane-F.molを使用します。
【配座探索計算の実行】
[Interfaceから実行する場合]
Cyclohexane-F.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログの「Calculation Type:」のプルダウンメニューから「Confromation Search」を選択します。「Search Limit:」の値は、「1.0」のままとします。これは、配座探索空間の範囲を決めます。この計算が終了した場合、最安定配座から1.0 kcal/molまでの領域が網羅されたことになります。
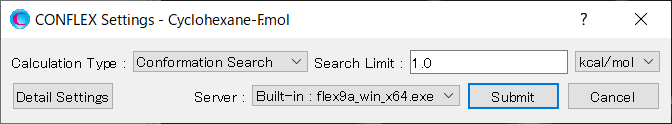
設定が終わりましたら、をクリックすると計算が始まります。
[コマンドラインから実行する場合]
計算設定は、Cyclohexane-F.iniファイルにキーワードを記述することで行います。
Cyclohexane-F.iniファイル
MMFF94S CONFLEX SEARCH=ENERGY SEL=1.0
それぞれのキーワードの意味は、次の通りです。
- MMFF94S
- MMFF94s力場を使う
- CONFLEX
- 配座空間探索を行なう
- SEARCH=ENERGY
- 配座空間の探索上限値は、エネルギー値で決める
- SEL=1.0
- 配座探索のエネルギー上限値は、1.0 kcal/molとする
なお、「MMFF94S」、「SEACH=ENERGY」、「SEL=1.0」はCONFLEXのデフォルト設定と等価なので、下記のiniファイルでも同じ計算が実行されます。
Cyclohexane-F.iniファイル
CONFLEX
Cyclohexane-F.molとCyclohexane-F.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。この計算が終了した場合、最安定配座から1.0 kcal/molまでの領域が網羅されたことになります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par Cyclohexane-Fenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
【配座探索計算の出力ファイル】
配座探索計算では、構造最適化計算の時と同じ3つの出力ファイル(Cyclohexane-F-F.mol、 Cyclohexane-F.bsf、 Cyclohexane-F.bso)の他に、以下のファイルが新たに作られます。
- Cyclohexane-F.fxf
- CONFLEXの配座探索計算において見出された配座が、このファイルに蓄えられます。このためCONFLEXでは最初に実行した際に作成された再実行ファイルを利用し、拡張された配座空間のみを探索することができます。なおこのファイルは、再度配座空間の探索を行なう時に必要です。
- Cyclohexane-F.fxo
- CONFLEXの配座探索計算において、CONFLEXが認識した分子の定義、Flip-Flap等の定義、そして配座探索において既に見出された配座との関係に関する検索結果が出力されます。
- Cyclohdt-F.ls1
- CONFLEXの配座探索計算において見出された配座リストが、立体エネルギーの安定な順に出力されます。
- Cyclohexane-F.ls2
- 配座探索計算において、得られた配座が新しいものであるかどうかの判定に用いた、2面角の値が出力されます。
- Cyclohexane-F.ls3
- 配座探索計算により得られた配座の座標データが出力されます。このファイルは、CONF. ID、立体エネルギー、立体エネルギーに基づく配座分布、配座の座標と原子リストから構成されます。
- Cyclohexane-F.ls4
- .ls1ファイルと同様に配座リストが出力されます。.ls1では立体エネルギーの安定な順に出力されていますが、.ls4では自由エネルギーの順に並べられています。
- Cyclohexane-F.sdf
-
CONFLEXの配座探索計算において見出された配座が、MDL-MOL形式の座標データで出力されます。
ファイル形式の詳細は、Cyclohexane.molファイルの内容に関する説明をご覧下さい。SDFファイルは、MOLファイルとは異なり、複数の分子構造定義が「$$$$」で区切られ出力されています。そのためSDFファイルを、「$$$$」をターゲットにしてファイル分割すれば、それぞれが分子構造の定義ファイル(MOLファイル)となります。このファイルの分子構造の定義ファイルの並び方は、全体のGibbsの自由エネルギーに従っています。ファイルの番号については、「INF=」がターゲットであり、前の部分が全体のGibbsの自由エネルギーに従った番号で、後の部分はCONFLEXが配座探索途中で見出した重複のない配座番号です。
0.7343750
18
2
1.00000000000000
5.200
1
T
F
0
F
6
D3D
-3.56093559779315
3.249848646468316E-009
0.888728442903559 1.48121407150593 39.2016730029237
-10.2067647343158 4.96801633911096 0.888728442903559
0.888728442903559 22.7814526607543 -5.90356166790034
2.98080980346657 106.763590387419 106.763590387419
8.44981392719261 104.244278365026 16.4665174320397
-3.56093559779315 -3.56093559779315 0.000000000000000E+000
-3.56093559779315 0.000000000000000E+000 0.000000000000000E+000
0.000000000000000E+000 0.000000000000000E+000 0.000000000000000E+000
0.000000000000000E+000 104.980111675433 105.572597304035
70.4329395908707 84.5730163650170 24.4153435746172
105.050542904982 13.6944155520592 0.000000000000000E+000
56.7385240388114 6.00000000000000
116.679654042328 116.679654042695 204.596311457385
0.000000000000000E+000 0.000000000000000E+000
***** START LOOP OF CONFLEX SEARCH ***** 18 TRIAL STRUCUTRES GENERATED FROM 1 INITIAL STRUCTURE(S). COUNT OF STACKED INITIAL STRUCUTRES: 1 INITIAL STRUCTURE - IDN: 1, EGY: -3.5609 NUMBER OF TRIAL CONFORMATIONS GENERATED: 18 NUMBER OF INITIAL STRUCTURES LEFT: 1 A NEW CONFORMER - IFT: 21,IDN: 2, EGY: 2.3688, GRMS: 0.81E-07, TIME: 0.05 REPLACED OLD ONE - IFT: 22,IDN: 2, EGY: 2.3688, GRMS: 0.50E-08, TIME: 0.03 IDENTICAL OLD ONE - IFT: 23,IDN: 2, EGY: 2.3688, GRMS: 0.74E-07, TIME: 0.03 REPLACED OLD ONE - IFT: 24,IDN: 2, EGY: 2.3688, GRMS: 0.98E-07, TIME: 0.05 IDENTICAL OLD ONE - IFT: 25,IDN: 2, EGY: 2.3688, GRMS: 0.64E-06, TIME: 0.03 IDENTICAL OLD ONE - IFT: 26,IDN: 2, EGY: 2.3688, GRMS: 0.17E-07, TIME: 0.05 IDENTICAL OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.14E-06, TIME: 0.03 REPLACED OLD ONE - IFT: 20,IDN: 1, EGY: -3.5609, GRMS: 0.32E-08, TIME: 0.03 REPLACED OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.20E-07, TIME: 0.05 IDENTICAL OLD ONE - IFT: 20,IDN: 1, EGY: -3.5609, GRMS: 0.14E-07, TIME: 0.05 IDENTICAL OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.73E-07, TIME: 0.03 IDENTICAL OLD ONE - IFT: 20,IDN: 1, EGY: -3.5609, GRMS: 0.14E-07, TIME: 0.05 IDENTICAL OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.17E-08, TIME: 0.03 IDENTICAL OLD ONE - IFT: 20,IDN: 1, EGY: -3.5609, GRMS: 0.11E-06, TIME: 0.03 IDENTICAL OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.37E-08, TIME: 0.03 IDENTICAL OLD ONE - IFT: 20,IDN: 1, EGY: -3.5609, GRMS: 0.15E-08, TIME: 0.08 IDENTICAL OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.26E-09, TIME: 0.03 IDENTICAL OLD ONE - IFT: 20,IDN: 1, EGY:
!====================================================================================!
! !
! LIST OF CONFORMERS (LIST FORM 1) !
! !
!------------------------------------------------------------------------------------!
! !
! DATE: 2020/09/04 TIME: 16:04:19.46 !
! Cyclohexane-F: Cyclohexane.mol !
! EMPIRICAL FORMULA: C6H12 MW = 84.094 !
! FORCE FIELD: MMFF94S(2010-12-04HG) !
! TOTAL NUMBER OF CONFORMERS FOUND: 2 !
! STERIC ENERGY: MIN= -3.5609 MAX= 2.3688 AVERAGE= -3.5607 !
! !
!------------------------------------------------------------------------------------!
! !
! DEFINITIONS: !
! NO. = INDEX NUMBER IN THE ORDER OF STERIC ENERGY !
! CONF. ID = IDENTIFICATION NUMBER OF EACH CONFORMER !
! STERIC E = STERIC ENERGY OF EACH CONFORMER (KCAL/MOL) !
! DELTA E = RELATIVE STERIC ENERGY FROM THE GLOBAL ENERGY MINIMUM (KCAL/MOL) !
! DISTRIBUTION = DISTRIBUTION BASED ON STERIC ENERGY (%) !
! INIT. = FLAG OF THE INITIAL STRUCTURE THAT HAD BEEN ALREADY USED !
! "*" = INDICATES THE INITIAL STRUCTURE USED IN THIS JOB !
! REOPT. = FLAG OF THE OPTIMIZED STRUCTURE THAT HAD BEEN OPTIMIZED AGAIN !
! "+" = INDICATES THE NEW CONFORMERS FOUND IN THIS JOB !
! NO. NEG. = NUMBER OF NEGATIVE EIGEN VALUES OF EACH CONFORMER !
! !
!====================================================================================!
====================================================================================
CONF. ORIGINAL DISTRI- NO.
NO. ID STERIC E DELTA E BUTION INIT. REOPT. NEG.
------------------------------------------------------------------------------------
1 00000001 -3.5609 0.0000 99.9955 T* F+ 0
2 00000002 2.3688 5.9297 0.0045 F F+ 0
------------------------------------------------------------------------------------
MINIMUM ENERGY: -3.5609 KCAL/MOL
AVERAGE ENERGY: -3.5607 KCAL/MOL (BASED ON THE BOLTZMANN LAW)
====================================================================================
各項目の意味は下記の通りです。
| キーワード | 説明 |
|---|---|
| NO. | 立体エネルギーの安定な順位 |
| CONF.ID | 探索により見出された順に付けられた番号 |
| ORIGINAL STERIC E | 立体エネルギー(kcal/mol) |
| DELTA E | 最安定配座からの立体エネルギー差(kcal/mol) |
| DISTRIBUTION | 立体エネルギーを元にBoltzmann分布により求めた配座分布 |
| INIT. | 初期構造として使用したかどうかを示すフラグ |
| REOPT. | すでに構造最適化が為されているか、またはこの配座探索計算で構造最適化を行ったかどうかを示すフラグ |
| NO. NEG. | 基準振動解析により得られた負の固有値の数 |
CONFLEXが配座探索時に初期配座として使用したものには、INIT.列に「T」と「*」で示します。「T」 は初期配座として既に用いたという意味であり、「*」はこの配座探索計算において初期配座として用いたことを意味します。
またCONFLEXでの配座探索中の構造最適化については、「F」と「+」で示します。「F」は構造最適が既に行われたことを意味し、 「+」 はこの配座探索計算において構造最適化をした新規配座であることを意味します。
またNO. NEGは、各配座における基準振動計算を行ない、その固有値が正であれば "0" で示し、その固有値が負であれば "1"で示されます。固有値が負を示す場合には、その分子における配座間を結ぶ重要な遷移状態を示すと考えられます。
なお、CONFLEXで得られた遷移状態については正確を期すために、必ず分子軌道法で遷移状態における構造最適化、基準振動解析及び固有反応座標計算による遷移状態の確認をすることをおすすめします。
!------------------------------------------------------------------------------------!
! !
! DEFINITIONS OF TORSION ANGLES: !
! D 1 = 3 - 1 - 2 - 16 !
! D 2 = 1 - 2 - 16 - 13 !
! D 3 = 2 - 16 - 13 - 8 !
! D 4 = 16 - 13 - 8 - 3 !
! D 5 = 13 - 8 - 3 - 1 !
! D 6 = 8 - 3 - 1 - 2 !
! !
!====================================================================================!
============================================================================================================
NO. ID STERIC E DELTA E DISTRIB. D 1 D 2 D 3 D 4 D 5 D 6
------------------------------------------------------------------------------------------------------------
1 00000001 -3.5609 0.0000 99.9955 -54.26 54.26 -54.26 54.26 -54.26 54.26
2 00000002 2.3688 5.9297 0.0045 29.17 29.17 -60.11 29.17 29.17 -60.11
------------------------------------------------------------------------------------------------------------
AVERAGE: -3.5607 0.0003
============================================================================================================
00000001 E= -3.5609; P= 99.9955 18 120. -1.26267 0.72901 0.22578 1 -0.00000 1.45801 -0.22578 1 -1.26267 -0.72901 -0.22578 1 -2.14619 1.23910 -0.17419 5 -1.33651 0.77163 1.31946 5 0.00000 2.47821 0.17419 5 -0.00000 1.54327 -1.31946 5 -0.00000 -1.45801 0.22578 1 -2.14619 -1.23910 0.17419 5 -1.33651 -0.77163 -1.31946 5 -0.00000 -2.47821 -0.17419 5 -0.00000 -1.54327 1.31946 5 1.26267 -0.72901 -0.22578 1 2.14619 -1.23910 0.17419 5 1.33651 -0.77163 -1.31946 5 1.26267 0.72901 0.22578 1 1.33651 0.77163 1.31946 5 2.14619 1.23910 -0.17419 5 STERIC E.= -3.56094 T F 00000002 E= 2.3688; P= 0.0045 18 120. -0.67151 1.22910 0.35868 1 -1.50939 0.00000 0.00000 1 0.67151 1.22910 -0.35868 1 -0.49926 1.24725 1.44233 5 -1.23203 2.13933 0.11739 5 -2.16866 0.23360 -0.84486 5 -2.16866 -0.23360 0.84486 5 1.50939 -0.00000 0.00000 1 0.49926 1.24725 -1.44233 5 1.23203 2.13933 -0.11739 5 2.16866 0.23360 0.84486 5 2.16866 -0.23360 -0.84486 5 0.67151 -1.22910 0.35868 1 1.23203 -2.13933 0.11739 5 0.49926 -1.24725 1.44233 5 -0.67151 -1.22910 -0.35868 1 -0.49926 -1.24725 -1.44233 5 -1.23203 -2.13933 -0.11739 5 STERIC E.= 2.36879 F F
!====================================================================================!
! !
! LIST OF CONFORMERS (LIST FORM 4) !
! !
!------------------------------------------------------------------------------------!
! !
! DATE: 2020/09/04 TIME: 16:04:19.46 !
! Cyclohexane-F: Cyclohexane.mol !
! EMPIRICAL FORMULA: C6H12 MW = 84.094 !
! FORCE FIELD: MMFF94S(2010-12-04HG) !
! TOTAL NUMBER OF CONFORMERS FOUND: 2 !
! STERIC ENERGY: EMIN= -3.5609 EMAX= 2.3688 EAVE= -3.5607 !
! GIBB'S FREE ENERGY: GMIN= 84.5730 GMAX= 88.7989 GAVE= 84.5764 !
! MIXING ENERGIES: HMIX= 105.5770 TSMIX= 21.0044 GMIX= 84.5725 !
! MIXING HEAT CAPACITY: CPMIX= 24.4158 !
! TEMPERATURE: 25.00 CELSIUS ( 298.15 KELVIN) !
! !
!------------------------------------------------------------------------------------!
! !
! DEFINITIONS: !
! NO. = INDEX NUMBER IN THE ORDER OF THE TOTAL GIBBS'S FREE ENERGY !
! CONF. ID = IDENTIFICATION NUMBER OF EACH CONFORMER !
! HE = ENTHALPY CONTRIBUTION OF EACH CONFORMER (KCAL/MOL) !
! TSE = ENTROPY (MULRPLIED BY TEMP.) CONTRIBUTION OF EACH CONFORMER (KCAL/MOL)!
! GE = TOTAL GIBB'S FREE ENERGY OF EACH CONFORMER (KCAL/MOL) !
! DGE = RELATIVE GIBB'S FREE ENERGY OF EACH CONFORMER (KCAL/MOL) !
! GEPOP = DISTRIBUTION IN GIBB'S FREE ENERGY (%) !
! SE = STERIC ENERGY OF EACH CONFORMER (KCAL/MOL) !
! DSE = RELATIVE STERIC ENERGY FROM THE GLOBAL ENERGY MINIMUM (KCAL/MOL) !
! SEPOP = DISTRIBUTION BASED ON STERIC ENERGY (%) !
! CHIRAL = CHIRALITY EXISTANCE OF EACH CONFORMER !
! SYM. NUM. = SYMMETRY NUMBER OF EACH CONFORMER !
! POINT GROUP = POINT GROUP OF SYMMETRY OF EACH CONFORMER !
! INIT. = FLAG OF EACH CONFORMER THAT HAD USED FOR INITIAL STRUCTURE !
! REOPT. = FLAG OF EACH CONFORMER THAT HAD BEEN OPTIMIZED AGAIN !
! NO. NEG. = NUMBER OF NEGATIVE EIGEN VALUES OF EACH CONFORMER !
! !
!====================================================================================!
========================================================================================================================================
CONF. SYM. POINT NO.
NO. ID HE TSE GE DGE GPOP SE DSE EPOP CHIRAL NUM. GROUP INIT. REOPT. NEG.
----------------------------------------------------------------------------------------------------------------------------------------
1 00000001 105.5726 20.9996 84.5730 0.0000 99.9202 -3.5609 0.0000 99.9955 F 6 D3D T F 0
2 00000002 111.0426 22.2436 88.7989 4.2259 0.0798 2.3688 5.9297 0.0045 T 4 D2 F F 0
----------------------------------------------------------------------------------------------------------------------------------------
========================================================================================================================================
各項目の意味は下記の通りです。
| キーワード | 説明 |
|---|---|
| NO. | 立体エネルギーの安定な順位 |
| CONF.ID | 探索により見出された順に付けられた番号 |
| HE | エンタルピー(kcal/mol) |
| TSE | エントロピーと温度の積(kcal/mol) |
| GE | Gibbsの自由エネルギー |
| DGE | 最安定配座からのGibbs自由エネルギー差(kcal/mol) |
| GPOP | Gibbs自由エネルギーを元にBoltzmann分布により求めた配座分布 |
| SE | 立体エネルギー(kcal/mol) |
| DSE | 最安定配座からの立体エネルギー差(kcal/mol) |
| EPOP | 立体エネルギーを元にBoltzmann分布により求めた配座分布 |
| CHIRAL | キラリティーを有するかどうかのフラグ |
| SYM. NUM. | 対称数 |
| POINT GROUP | 点群の記号 |
| INIT. | 初期構造として使用したかどうかを示すフラグ |
| REOPT. | すでに構造最適化が為されているか、またはこの配座探索計算で構造最適化を行ったかどうかを示すフラグ |
| NO. NEG. | 基準振動解析により得られた負の固有値の数 |
Cyclohexane.mol
CONFLEX 20090416043D 1 1.00000 -3.56094 1
D3D ,E = -3.561, G = 0.325E-08, P = 99.9955, M( 0), IFN =00000001-00000001
18 18 0 0 999 V2000
-1.2627 0.7290 0.2258 C 0 0 0 0 0
-0.0000 1.4580 -0.2258 C 0 0 0 0 0
-1.2627 -0.7290 -0.2258 C 0 0 0 0 0
-2.1462 1.2391 -0.1742 H 0 0 0 0 0
-1.3365 0.7716 1.3195 H 0 0 0 0 0
0.0000 2.4782 0.1742 H 0 0 0 0 0
-0.0000 1.5433 -1.3195 H 0 0 0 0 0
-0.0000 -1.4580 0.2258 C 0 0 0 0 0
-2.1462 -1.2391 0.1742 H 0 0 0 0 0
-1.3365 -0.7716 -1.3195 H 0 0 0 0 0
-0.0000 -2.4782 -0.1742 H 0 0 0 0 0
-0.0000 -1.5433 1.3195 H 0 0 0 0 0
1.2627 -0.7290 -0.2258 C 0 0 0 0 0
2.1462 -1.2391 0.1742 H 0 0 0 0 0
1.3365 -0.7716 -1.3195 H 0 0 0 0 0
1.2627 0.7290 0.2258 C 0 0 0 0 0
1.3365 0.7716 1.3195 H 0 0 0 0 0
2.1462 1.2391 -0.1742 H 0 0 0 0 0
2 1 1 0 0
3 1 1 0 0
1 4 1 0 0
1 5 1 0 0
2 6 1 0 0
2 7 1 0 0
2 16 1 0 0
3 8 1 0 0
3 9 1 0 0
3 10 1 0 0
11 8 1 0 0
12 8 1 0 0
13 8 1 0 0
14 13 1 0 0
13 15 1 0 0
16 13 1 0 0
16 17 1 0 0
16 18 1 0 0
M END
> <DATE_YYYY/MM/DD>
2020/09/04
> <TIME_HH:MM:SS.XX>
16:04:19.46
> <MOL_FILE_NAME>
Cyclohexane-F: Cyclohexane.mol
> <FORCE_FIELD_NAME>
MMFF94S(2010-12-04HG)
> <TEMPERATURE_K>
298.15
> <TOTAL_NUMBER_OF_CONFORMERS>
2
> <CONFORMER_ID>
00000001
> <POTENTIAL_ENERGY_KCAL/MOL>
-3.560936
> <ENERGY_RMS_GRADIENT_KCAL/MOL/ANGS>
3.2500000E-09
> <BOLTZMANN_POPULATION_%>
99.99550
> <TOTAL_GIBBS_FREE_ENERGY_KCAL/MOL>
84.57302
> <TOTAL_ENTHALPY_KCAL/MOL>
105.5726
> <TOTAL_ENTROPY_CAL/MOL/K>
70.43294
> <HEAT_CAPACITY_CAL/MOL/K>
24.41534
> <ENTHALPY_FUNCTION_CAL/MOL/K>
13.69442
> <FREE_ENERGY_FUNCTION_CAL/MOL/K>
56.73852
> <CHIRALITY>
F
> <SYMMETRY_NUMBER>
6
> <POINT_GROUP>
D3D
> <NUMBER_OF_NEGATIVE_EIGENVALUES>
0
> <CONFLEX_PERTURBATION_FLAG>
T
> <CONFLEX_REOPTIMIZATION_FLAG>
F
$$$$
【配座探索空間の拡張】
前節のCyclohexane-F.ls1を見ますと、2番目の配座のINIT.の列が「F」になっています。これは、この構造がまだ配座探索の初期構造として使用されていないという意味で、この構造から派生する配座異性体はまだ得られていない可能性があります。
そこで、この構造を初期構造として探索するために、配座探索空間を拡張して再度計算を行います。
[Interfaceから実行する場合]
Cyclohexane-F.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログの「Calculation Type:」のプルダウンメニューから「Confromation Search」を選択します。「Search Limit:」の値は、「6.0」に変更します。これは、最安定構造と2番目の配座のエネルギー差が5.9297kcal/molであるためです。この設定により、2番目の配座が探索の初期構造として利用されるようになります。
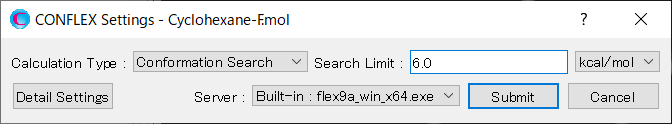
設定が終わりましたら、をクリックします。計算が始まります。
この計算が終了した場合、最安定配座から6.0 kcal/molまでの領域が網羅されたことになります。
なお、配座探索空間を拡張して再計算を行う場合は、最初の計算時に出力されたCyclohexane-F.fxfファイルが、Cyclohexane-F.molと同じフォルダに格納されていなければなりません。
[コマンドラインから実行する場合]
Cyclohexane-F.iniファイルの「SEL=」の値を「6.0」に変更します。
これは、最安定構造と2番目の配座のエネルギー差が5.9297kcal/molであるためです。この設定により、2番目の配座が探索の初期構造として利用されるようになります。
Cyclohexane-F.iniファイル
MMFF94S CONFLEX SEACH=ENERGY SEL=6.0
Cyclohexane-F.molとCyclohexane-F.ini、Cyclohexane-F.fxfの三つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
この計算が終了した場合、最安定配座から6.0 kcal/molまでの領域が網羅されたことになります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par Cyclohexane-Fenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
計算後に出力されたCyclohexane-F.ls1ファイルを確認すると、2番目の配座の「INIT.」値が「T」に変わっていることがわかります。つまり、2番目の配座が探索の初期構造として利用されたことを意味します。
====================================================================================
CONF. ORIGINAL DISTRI- NO.
NO. ID STERIC E DELTA E BUTION INIT. REOPT. NEG.
------------------------------------------------------------------------------------
1 00000001 -3.5609 0.0000 99.9955 T F 0
2 00000002 2.3688 5.9297 0.0045 T* F 0
------------------------------------------------------------------------------------
MINIMUM ENERGY: -3.5609 KCAL/MOL
AVERAGE ENERGY: -3.5607 KCAL/MOL (BASED ON THE BOLTZMANN LAW)
====================================================================================
再計算の結果、新たな配座は得られなかったので、CONFLEXのアルゴリズムで探索できる範囲内での、MMFF94s力場によるエネルギー極小構造は全て網羅したことになります。
このように、 ls1ファイルの情報を元に、 SELの値を徐々に大きくして計算を繰り返すことで、探索する配座空間領域を広げることができます。全ての配座異性体を初期構造として使用し、新規の構造が得られなくなった時点で、適用した力場による配座空間を完全に網羅したと見なすことができます。
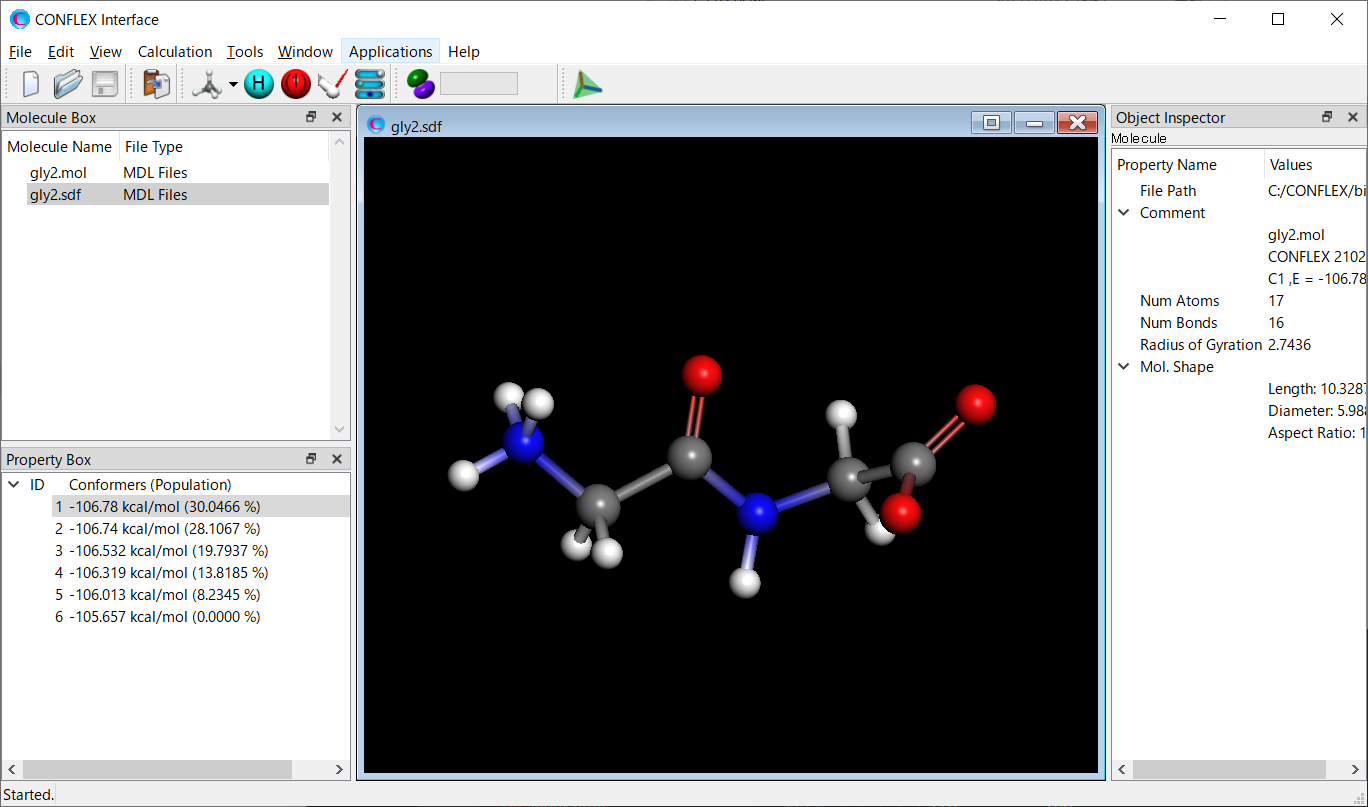
【計算結果の可視化】
[Interfaceから実行した場合]
計算の実行後、CONFLEX Interfaceの下部にJob Managerが現れます。Job Managerには、実行した計算の状態が表示されます。
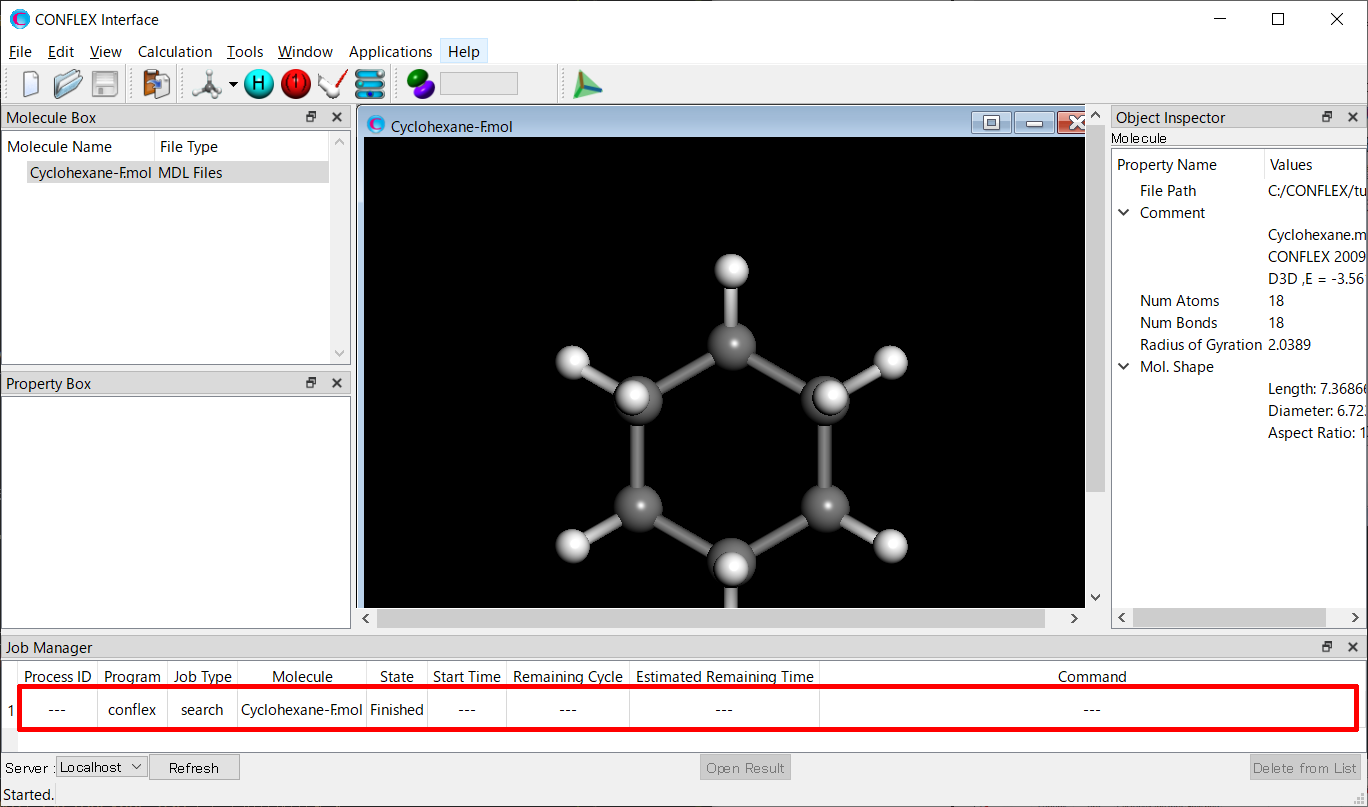
配座探索計算を行ったJobのStateがFinishedであることを確認し、表示部分(赤枠部分)をダブルクリックしてください。Cyclohexane-F.sdfファイルが開き、配座異性体が描画されます。
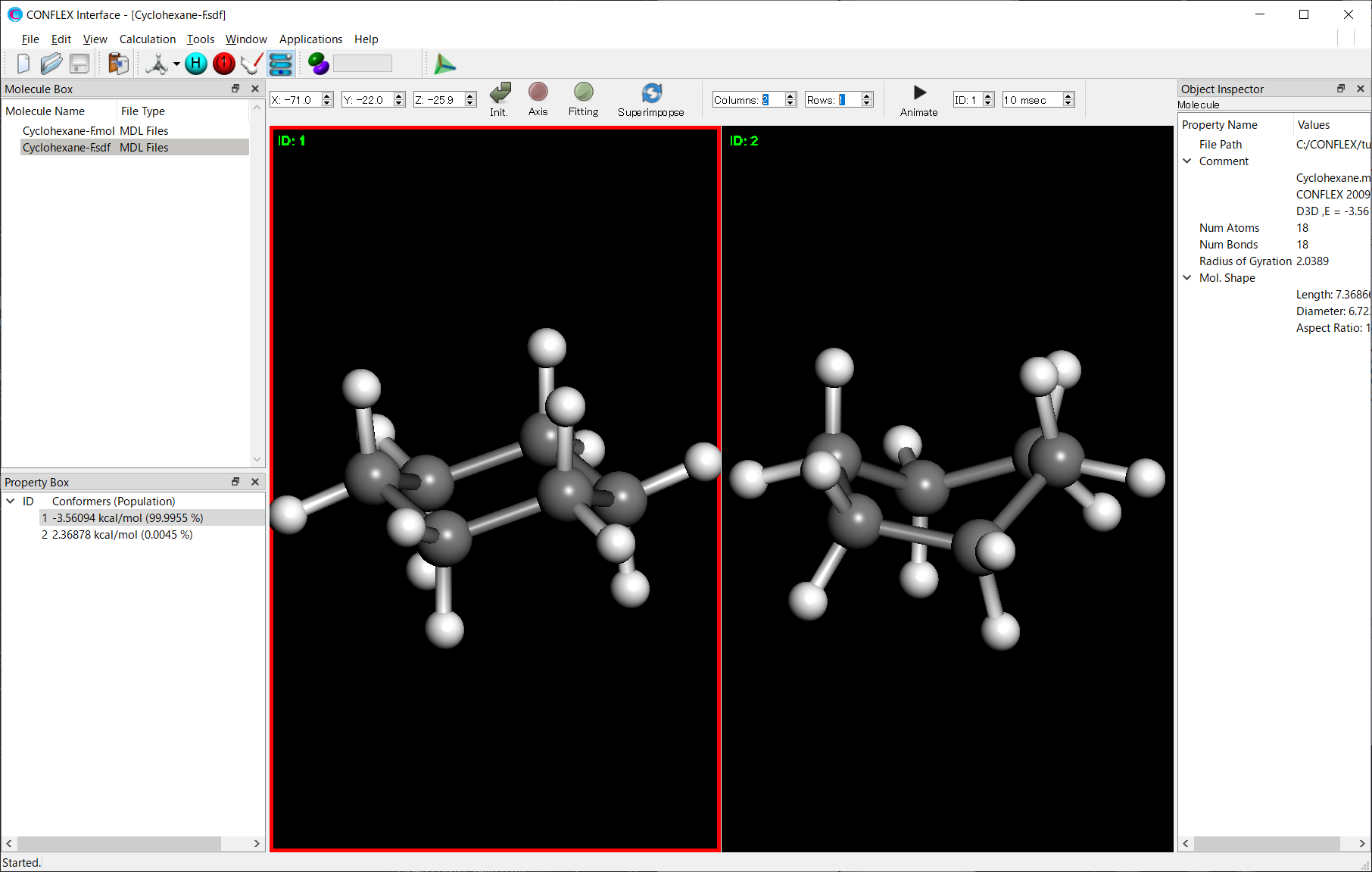
*画面分割は、Viewメニューの「Controller」を選択して表示されるツールバーの「Columns/Rows」を設定することで可能です。
[コマンドラインから実行した場合]
入力ファイルを格納したフォルダに、Cyclohexane-F.sdfファイルがあります。これをCONFLEX Interfaceで開くことで、配座異性体を可視化できます。
*画面分割は、Viewメニューの「Controller」を選択して表示されるツールバーの「Columns/Rows」を設定することで可能です。
【溶媒効果を取り入れた探索】
GB/SAモデルにより溶媒効果を導入した配座探索を行う方法について説明します。
GB/SAモデルに関する説明は【溶媒効果を取り入れた計算】をご覧ください。
zwitter ion型のグリシン2量体を例にします。
zwitter ion型グリシン2量体の座標データ(gly2.mol)
gly2.mol
17 16 0 0 0 0 0 0 0 0999 V2000
-1.2219 0.9765 -2.1504 N 0 3 0 0 0 0 0 0 0 0 0 0
0.2781 0.9765 -2.1504 C 0 0 0 0 0 0 0 0 0 0 0 0
0.6496 2.0256 -2.1504 H 0 0 0 0 0 0 0 0 0 0 0 0
0.7803 0.2658 -0.9176 C 0 0 0 0 0 0 0 0 0 0 0 0
1.9679 0.1552 -0.7258 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.1098 -0.2537 -0.0165 N 0 0 0 0 0 0 0 0 0 0 0 0
0.3728 -0.9366 1.1681 C 0 0 0 0 0 0 0 0 0 0 0 0
0.9911 -0.2372 1.7741 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.8018 -1.4089 1.9892 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.6088 -2.0256 3.0592 O 0 0 0 0 0 0 0 0 0 0 0 0
-1.9679 -1.1835 1.5993 O 0 5 0 0 0 0 0 0 0 0 0 0
-1.5708 0.4839 -3.0034 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.5698 0.4832 -1.2973 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.5701 1.9618 -2.1504 H 0 0 0 0 0 0 0 0 0 0 0 0
0.6489 0.4518 -3.0592 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.1047 -0.1611 -0.1772 H 0 0 0 0 0 0 0 0 0 0 0 0
0.9910 -1.8114 0.8658 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
1 12 1 0
1 13 1 0
1 14 1 0
2 3 1 0
2 4 1 0
2 15 1 0
4 5 2 0
4 6 1 0
6 7 1 0
6 16 1 0
7 8 1 0
7 9 1 0
7 17 1 0
9 10 2 0
9 11 1 0
M END
[Interfaceから実行する場合]
gly2.molファイルをCONFLEX Interfaceを用いて開きます。
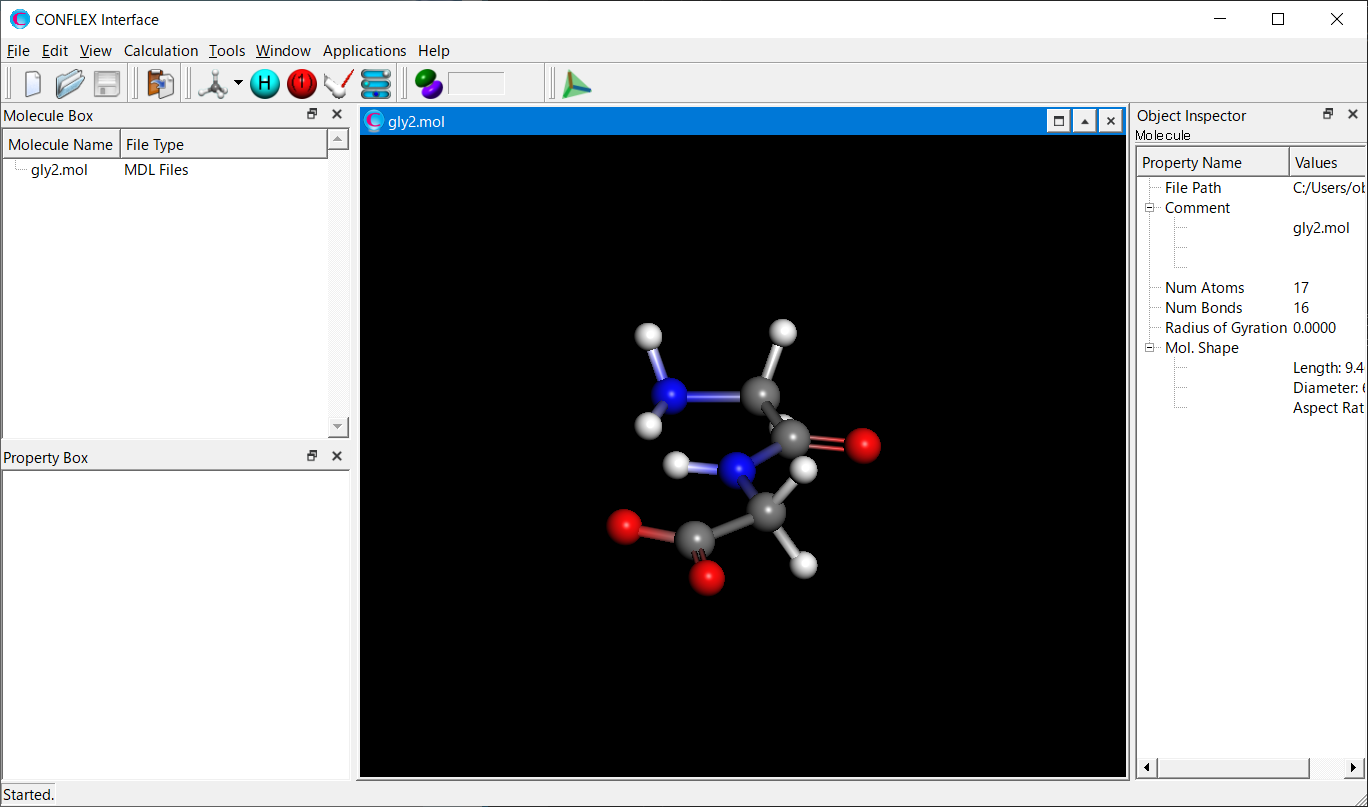
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。詳細設定ダイアログが表示されます。
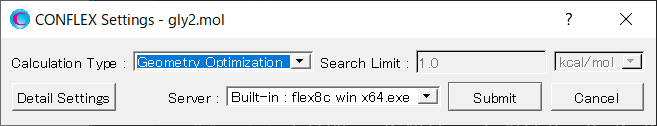
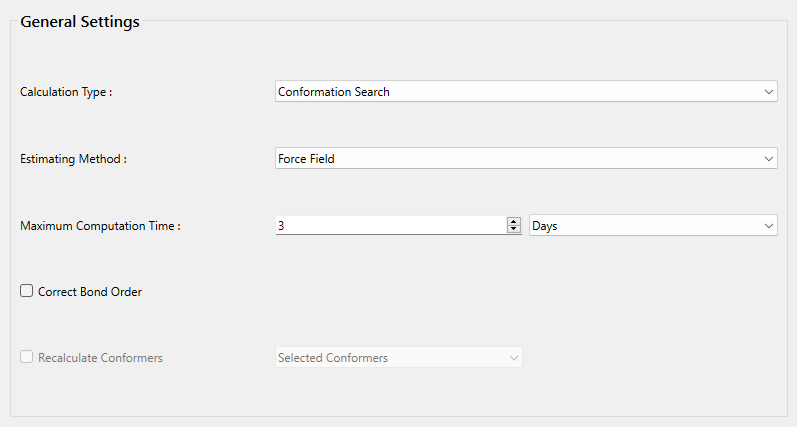
詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」 のプルダウンメニューから「Conforamtion Search」を選択します。
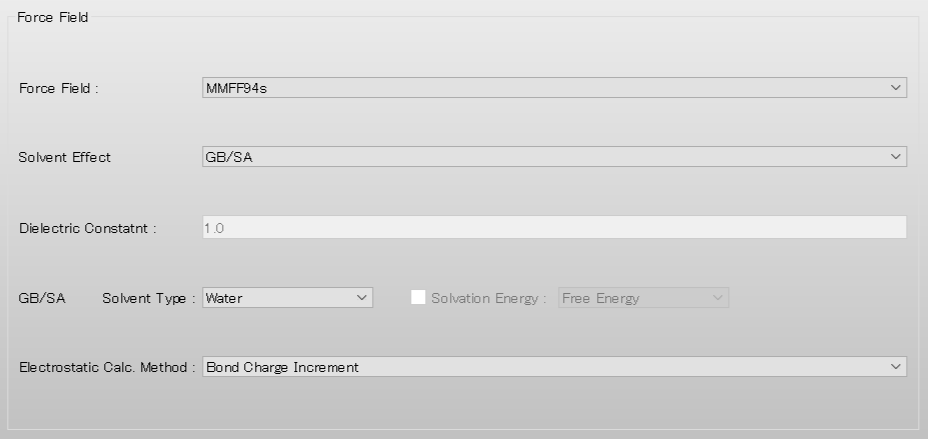
続いて、詳細設定ダイアログの「Force Field」ダイアログにある「Solvent Effect」 のプルダウンメニューから「GBSA」を選択します。
最後に、「Conformation Search」の「Search Limit:」の値を「10.0」に設定します。この計算が終了した場合、最安定配座から10.0 kcal/molまでの領域が網羅されたことになります。
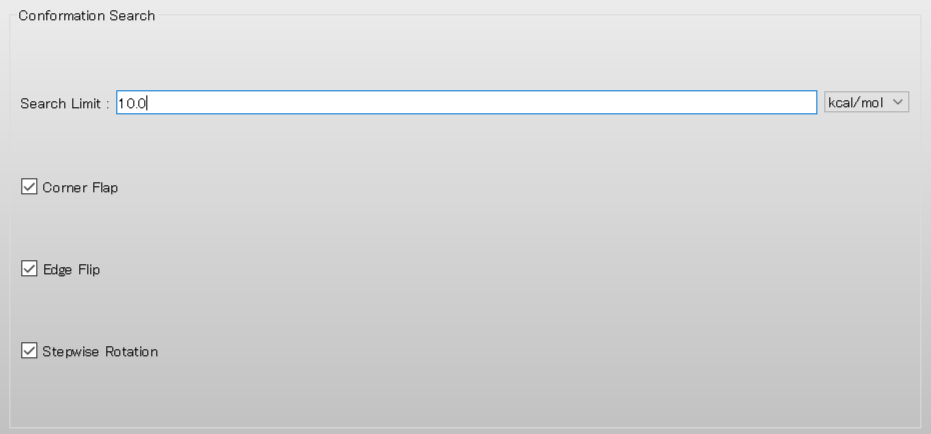
設定が終わりましたら、詳細設定ダイアログのをクリックします。溶媒効果を含む配座探索が行われます。
[コマンドラインから実行する場合]
計算設定は、gly2.iniファイルにキーワードを記述することで行います。
gly2.iniファイル
MMFF94S GBSA CONFLEX SEL=10.0
| キーワード | 説明 |
|---|---|
| MMFF94S | MMFF94s力場を用いて計算を行うことを意味します。 |
| GBSA | GB/SAモデルを導入した計算が実行できます。 |
| CONFLEX | 配座探索を実行することを意味します。 |
| SEL=10.0 | 配座探索空間の範囲。最安定配座から10.0 kcal/molまでの領域が網羅されます。 |
gly2.molとgly2.iniをフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par gly2enter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
最安定配座異性体の構造を下記に示します。可視化の方法は、「計算結果の可視化」をご覧ください。