Introduction
Thank you very much for purchasing CONFLEX DOCK.
CONFLEX DOCK is a docking simulation program that predicts where a specified peptide chain will coordinate with a substrate protein to form a complex.
When making predictions, the amino acid residues of the protein are coarse-grained at representative points (Cα atoms) and a tetrahedron is constructed by Delaunay partitioning. Then, peptide residues are placed at search points set on the protein surface, and scores are evaluated based on the tetrahedral coarse-grained potentials.
Citation
T. Yamamoto, Y. Ikabata, H. Goto, “Reconstruction of Four-Body Statistical Pseudopotential for Protein-Peptide Docking”, J. Comput. Chem., Jpn.-Int. Ed., 2024, 10, 2023-0039.
Execution procedure
Path to the executables
The CONFLEX DOCK executable, parameter files and the license file are installed in the following folder.
| OS | executable file | License Parameters |
|---|---|---|
| Windows | C:\CONFLEX\bin | C:\CONFLEX\par |
| macOS | /Applications/CONFLEX/bin | /Applications/CONFLEX/par |
| Linux | /usr/local/conflex/bin | /usr/local/conflex/par |
Input files
In CONFLEX DOCK, the structural data of the protein must be provided as a PDB file, and information about the peptide is also required for the calculation. The peptide information can be specified in either of the following ways:
- By providing a PDB file that contains the peptide structure data
-
By specifying the amino acid sequence using either:
- One-letter notation, or
- Three-letter notation.
The calculation settings are made by creating an ini file with the “.ini” extension, in which various keywords are specified. If CONFLEX DOCK is run from the CONFLEX Interface, the ini file is created automatically; however, if run from the command line, users must prepare the ini file themselves before running the calculation.
It is possible to run the calculation without an ini file, but in that case, the calculation will be performed using the default settings.
Execution from Interface
Start the CONFLEX Interface
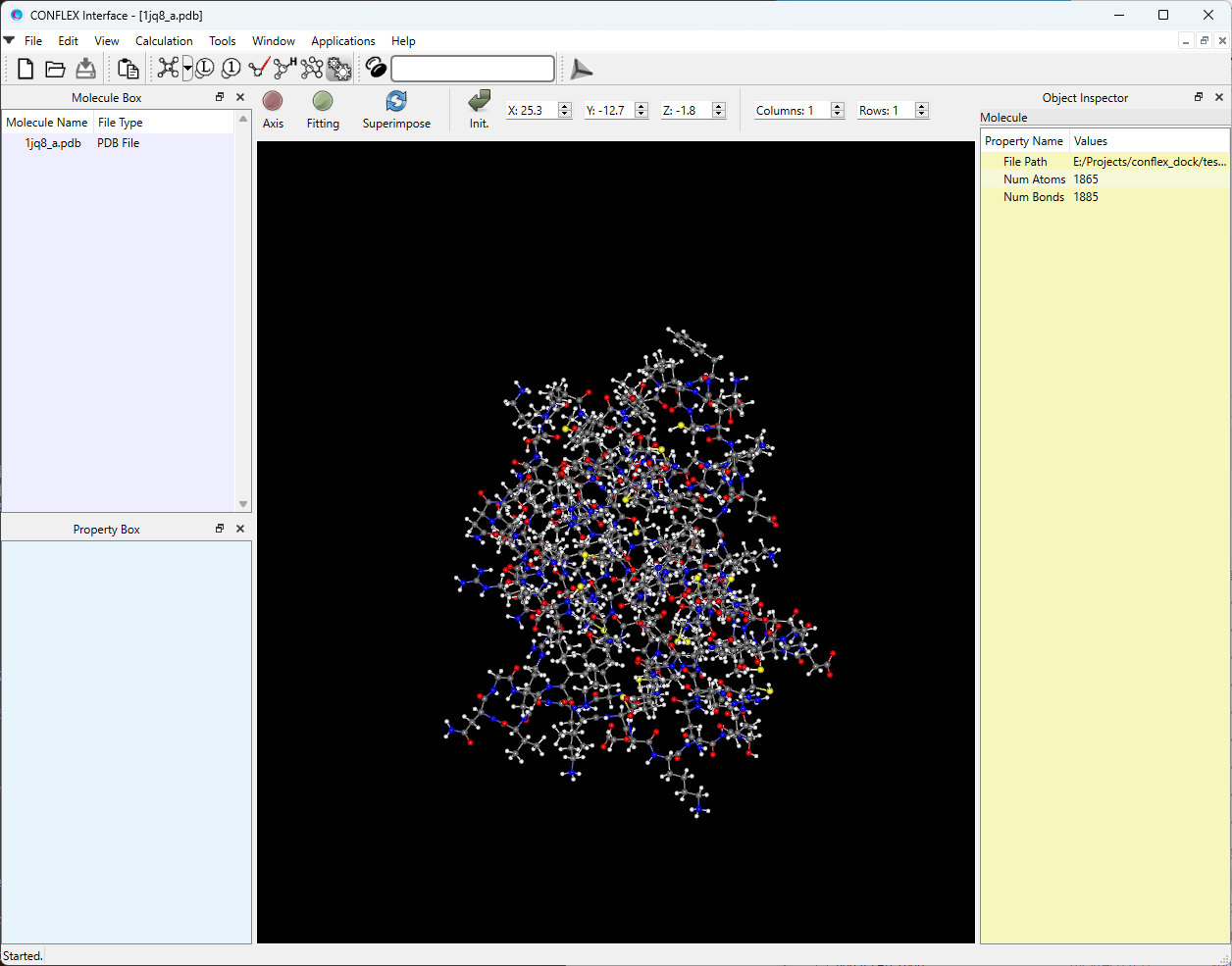
Next, open the PDB file of the protein.
You can either select “Open” from the File menu and specify the PDB file, or drag and drop the PDB file onto the gray area of the CONFLEX Interface.
The protein will then be displayed as shown below.
Next, select “Docking” from the Calculation menu.
This will open the calculation settings dialog as shown below:
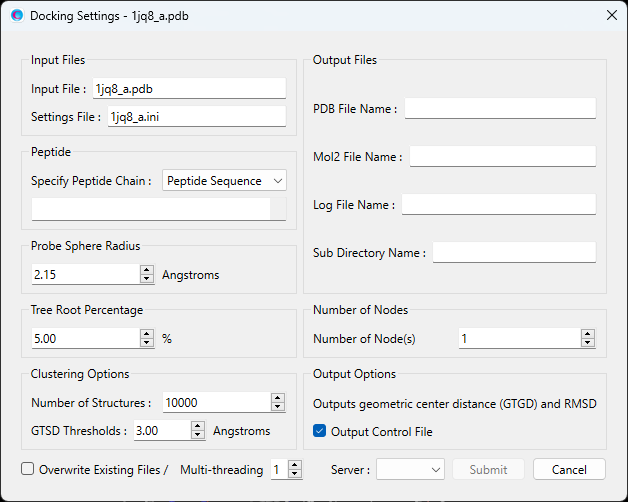
You can set the peptide information under the “Peptide” section.
Select “Peptide Sequence” from the dropdown menu, then enter the amino acid sequence in either one-letter or three-letter notation in the input box.
If using three-letter notation, separate each amino acid residue with a hyphen “-” (for example: ARG-ARG-ASP-TYR-PHE).
Alternatively, you can specify the peptide information using a PDB file by selecting “Open File” from the dropdown menu. Click the “Open File” button and select the appropriate PDB file.
In addition, you can set other calculation settings and specify output file names in this dialog.
Once the calculation settings are complete, click to start the calculation.
Execution from Command line
When running the calculation from the command line, use the “-par” option to specify the folder where the license and parameter files are stored. Use the “-ipro” option to specify the PDB file of the protein, and the “-ipep” option to specify either the peptide file name or the amino acid sequence.
Although the command is shown on multiple lines, please enter it as a single line.
Windows:
C:\CONFLEX\bin\conflex_dock-1a.exe -par C:\CONFLEX¥par
(-omp <number of parallel threads>)
-ipro <Protein file name> -ipep <Peptide file name or sequence>
(-ini <setup file name> -olog <log file name>
-opdb <Output PDB file name> -omol2 <Output Mol2 file name>
-odir <output folder name>)enter
macOS:
/Applications/CONFLEX/bin/conflex_dock-1a.exe -par /Applications/CONFLEX/par
(-omp <number of parallel threads>)
-ipro <Protein file name> -ipep <Peptide file name or sequence>
(-ini <setup file name> -olog <log file name>
-opdb <Output PDB file name> -omol2 <Output Mol2 file name>
-odir <output folder name>)enter
Linux:
/usr/local/conflex/bin/conflex_dock-1a.exe -par /usr/local/conflex/par
(-omp <number of parallel threads>)
-ipro <Protein file name> -ipep <Peptide file name or sequence>
(-ini <setup file name> -olog <log file name>
-opdb <Output PDB file name> -omol2 <Output Mol2 file name>
-odir <output folder name>)enter
The “-ipro” and “-ipep” options are required. You can also specify the output file name and the configuration file to be used with optional parameters.
The available options are listed below.
| Command line arguments | Description |
|---|---|
| -par |
Specify the directory containing the license and score files. If not specified, the current directory is set. |
| -omp <numerical value> *only in the Parallel ver. |
Specify the number of threads for parallel computation. |
| -ipro <File name> |
Specify the protein PDB file. If not specified, the calculation is stopped. |
|
-ipep <File name> -ipep <peptide sequence> |
Specify the PDB file or array (single or three letter notation) of the peptide. If not specified, the calculation is stopped. |
| -ini <File name> |
Specify the calculation setup file. If not specified, all calculation conditions are executed by default. |
| -olog <File name> |
Set the log file name. If not specified, the file name is set to a combination of the file name specified with -ipro and the file name or array specified with -ipep. |
| -opdb <File name> |
Set the PDB file name to output docking (clustering) results. If not specified, the file name is set to a combination of the file name specified with -ipro and the file name or array specified with -ipep. |
| -omol2 <File name> |
Set the mol2 file name to output docking results (by pose). If not specified, the file name is set by combining the file name specified with -ipro and the file name or array specified with -ipep. |
| -odir <directory name> |
Set the name of the directory to output csv files, etc. If not specified, the directory name is set to a combination of the file name specified with -ipro and the file name or array specified with -ipep. |
Output files
If you run
/Applications/CONFLEX/bin/conflex_dock-1a.exe -par /Applications/CONFLEX/par -ipro protein.pdb -ipep peptide.pdb
with the protein file named “protein.pdb” and the peptide file named “peptide.pdb”, the calculation results will be output with the following file names and folder names.
| File/Dir name | Description |
|---|---|
| protein_peptide.log | This file contains the calculation settings used, score values for each docking pose, their distribution, clustering results, and other related informations. |
| protein_peptide.pdb | This file contains each docking pose output in PDB format. |
| protein_peptide.mol2 | This file contains each docking pose represented by coarse-grained representative points in Mol2 format. |
| protein_peptide/ | This is the directory where the calculation results are output as CSV files. |
If the peptide information is specified using an amino acid sequence as shown below:
/Applications/CONFLEX/bin/conflex_dock-1a.exe -par /Applications/CONFLEX/par -ipro protein.pdb -ipep LAIYS
the output file name and the generated folder name will be “protein_LAIYS”. The output file name and folder name can be changed by using command options.