Amino acid residue substitution function
[Overview]
Some amino acid residues in proteins within the body may undergo mutations due to various influences. Even a protein with mutations in just a few residues can exhibit significant changes in function and activity. To study such phenomena through simulation, you can obtain a PDB file which contains the amino acid sequence of the protein from the Protein Data Bank (PDB), and then a mutated version of the protein is created using amino acid substitutions.
When using a PDB file obtained from the PDB, you may need to correct or complete missing side chain data for certain amino acids. CONFLEX allows you to replace or supplement these missing side chains in the PDB file before performing geometry optimization or other calculations. Additionally, CONFLEX provides information about missing data in the PDB file, whether it pertains to side chains or the backbone data.
[Substitution of side chain]
We use a PDB file that contains missing atoms, correct the data, and then perform a calculation.
First, search “1OHR” from PDB web site, and download 1ohr.pdb file.
This structure is an HIV-1 protease containing an inhibitor obtained by X-ray crystal structure analysis. It is a dimeric protein with 99 amino acid residues. In the 1ohr.pdb file, the lines starting with “REMARK 470” contain information about the missing atoms.
Lines starting with “REMARK 470” in the 1ohr.pdb file
REMARK 470 REMARK 470 MISSING ATOM REMARK 470 THE FOLLOWING RESIDUES HAVE MISSING ATOMS (M=MODEL NUMBER; REMARK 470 RES=RESIDUE NAME; C=CHAIN IDENTIFIER; SSEQ=SEQUENCE NUMBER; REMARK 470 I=INSERTION CODE): REMARK 470 M RES CSSEQI ATOMS REMARK 470 GLN A 7 CD OE1 NE2 REMARK 470 LYS A 14 CE NZ REMARK 470 GLU A 34 CD OE1 OE2 REMARK 470 GLU A 35 CD OE1 OE2 REMARK 470 ARG A 41 CG CD NE CZ NH1 NH2 REMARK 470 LYS A 43 CG CD CE NZ REMARK 470 LYS A 45 CG CD CE NZ REMARK 470 LYS A 55 CD CE NZ REMARK 470 GLN A 61 CG CD OE1 NE2 REMARK 470 LYS A 70 CE NZ REMARK 470 GLN B 7 CD OE1 NE2 REMARK 470 LYS B 14 CG CD CE NZ REMARK 470 ARG B 41 CG CD NE CZ NH1 NH2 REMARK 470 LYS B 55 CE NZ REMARK 470 GLN B 61 CD OE1 NE2
The line [REMARK 470 GLN A 7 CD OE1 NE2] indicates that the Cδ, Oε1, and Nε2 of glutamine residue at positon 7 in chain A are missing. CONFLEX uses lines starting with “ATOM” to retrieve structure data. Therefore, modifying the information in “REMARK 470” will not affect the calculation.
Execute CONFLEX using the 1ohr.pdb file.
[Execution from Interface]
Open the 1ohr.pdb file using CONFLEX Interface.
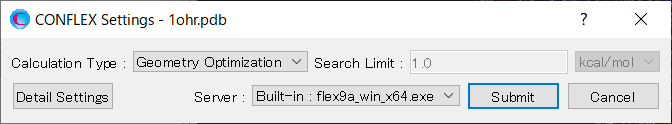
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears.
Next, click at the bottom Next, click at the bottom right of the detailed settings dialog.
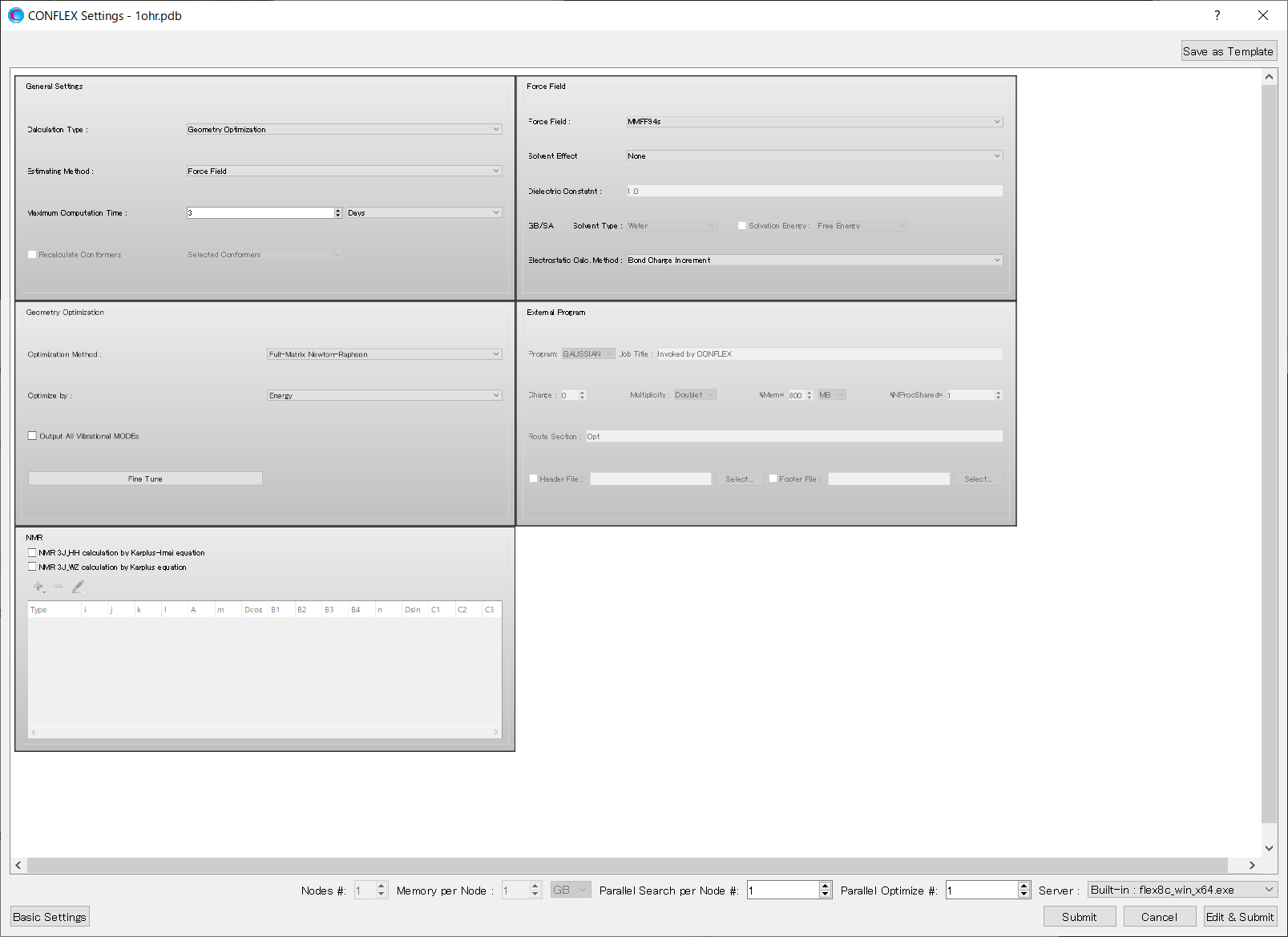
After clicking, a dialog displaying the keywords for the calculation settings will appear.
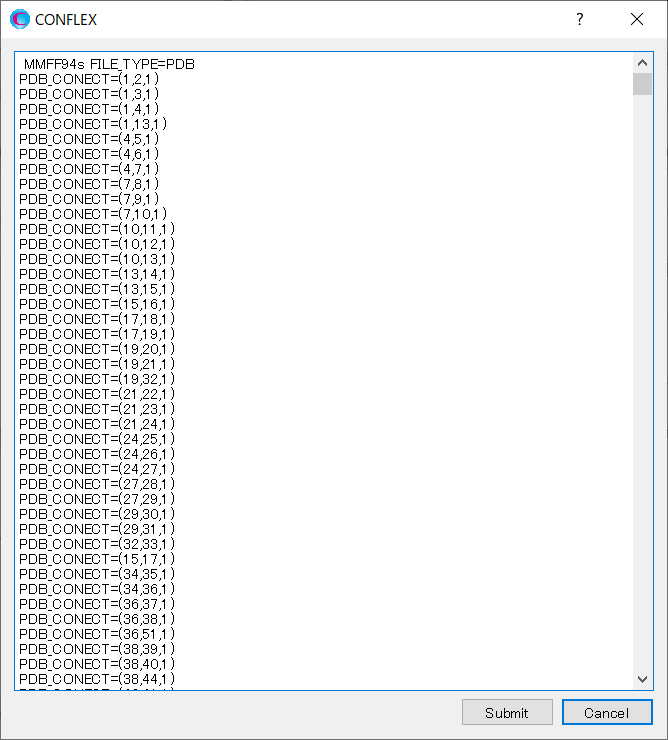
Delete all the “PDB_CONECT=” keywords automatically generated by Interface program, and add information of double bonds for the inhibitor using “PDB_CONECT=” keyword.
In “PDB_CONECT=(i,j,n)”, i and j are the serial number of atoms as described in the pdb file, and n represents the bond order.
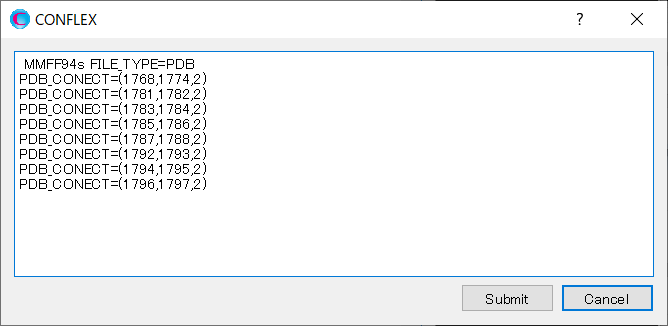 When completing the modifications, click to start the calculation.
When completing the modifications, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the 1ohr.ini file.
1ohr.ini file
MMFF94S PDB_CONECT=(1768,1774,2) PDB_CONECT=(1781,1782,2) PDB_CONECT=(1783,1784,2) PDB_CONECT=(1785,1786,2) PDB_CONECT=(1787,1788,2) PDB_CONECT=(1792,1793,2) PDB_CONECT=(1794,1795,2) PDB_CONECT=(1796,1797,2)
[MMFF94S] keyword means to use MMFF94s force field.
[PDB_CONECT=] keywords set information of double bonds for the inhibitor.
In “PDB_CONECT=(i,j,n)”, i and j are the serial number of atoms as described in the pdb file, and n represents the bond order.
Store the two files of 1ohr.pdb and 1ohr.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par 1ohrenter
The above command is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
When you check the bso file after the caluclation has finished, you will get the following messages. These messages indicate that errors occurred during the calculation. Confirm that the errors correspond to the missing atoms mentioned earlier.
Error messages in the bso file
PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 7 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLN,A,7) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 14 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,A,14) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 34 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLU,A,34) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 35 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLU,A,35) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 41 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(ARG,A,41) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 43 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,A,43) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 45 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,A,45) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 55 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,A,55) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 61 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLN,A,61) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 70 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,A,70) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN B 7 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLN,B,7) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN B 14 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,B,14) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN B 41 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(ARG,B,41) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN B 55 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,B,55) PDB_EXT/CHECK_AND_BUILD: ERROR -- INCLUDE UNKNOWN RESIDUE IN CHAIN B 61 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLN,B,61)
There are two options to avoid the errors and perform the calculation.
Execution using original coordinates
One of the options is to execute the calculation using the original coordinates.
In this case, add [PDB_NOMUTATE] keyword to the calculation settings.
[PDB_NOMUTATE] means to directly use the original data, including any missing atoms in the amino acid residues.
[Execution from Interface]
Add [PDB_NOMUTATE] keyword to the dialog that appears when you click .
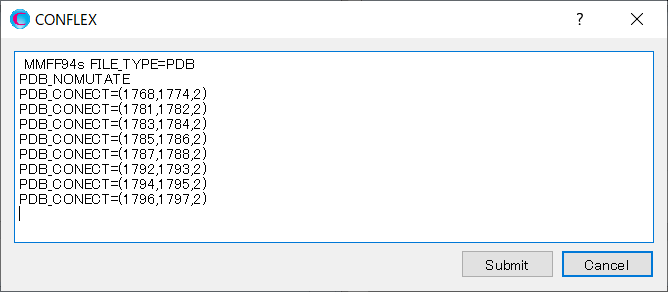 When completing the modification, click to start the calculation.
When completing the modification, click to start the calculation.
[Execution from command line]
Add [PDB_NOMUTATE] keyword to the 1ohr.ini file.
1ohr.ini file
MMFF94S
PDB_NOMUTATE
PDB_CONECT=(1768,1774,2)
PDB_CONECT=(1781,1782,2)
PDB_CONECT=(1783,1784,2)
PDB_CONECT=(1785,1786,2)
PDB_CONECT=(1787,1788,2)
PDB_CONECT=(1792,1793,2)
PDB_CONECT=(1794,1795,2)
PDB_CONECT=(1796,1797,2)
Store the two files of 1ohr.pdb and 1ohr.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par 1ohrenter
The above command is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
When you check the bso file after the caluclation has finished, you will get the following messages. You will notice that the error messages have been replaced with warning messages.
* Note that the calculation may take a long time to complete due to the large size of the molecule.
Warning messages in the bso file
PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 7 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLN,A,7) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 14 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,A,14) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 34 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLU,A,34) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 35 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLU,A,35) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 41 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(ARG,A,41) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 43 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,A,43) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 45 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,A,45) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 55 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,A,55) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 61 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLN,A,61) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN A 70 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,A,70) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN B 7 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLN,B,7) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN B 14 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,B,14) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN B 41 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(ARG,B,41) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN B 55 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(LYS,B,55) PDB_EXT/CHECK_AND_BUILD: WARNING -- INCLUDE UNKNOWN RESIDUE IN CHAIN B 61 *PLEASE SET KEYWORD(S) :PDB_MUTATE=(GLN,B,61)
Execution using substitution of side chain
The other option is to supplement the missing atoms and then perform the calculation. In the warning messages above, you will see the “PDB_MUTATE=” keywords. Delete the “PDB_NOMUTATE” keyword and add the “PDB_MUTATE=” keywords as shown above.
“PDB_MUTATE=” is a keyword used to replace or supplement a side chain. The first element in the parentheses is the amino acid name, the second is the protein chain ID, and the third is the residue number. The amino acid name can be represented in 1-character, 3-character, or full-character notation (see the table below). For example, “PDB_MUTATE=(GLN,A,7)” means replacing the 7th residue of chain A with glutamine.
| Name of amino acid | 1-Character | 3-Characters | Full name |
|---|---|---|---|
| Alanine | A | ALA | ALANINE |
| Cysteine | C | CYS | CYSTEINE |
| Aspartic acid | D | ASP | ASPARTIC_ACID |
| Glutamic acid | E | GLU | GLUTAMIC_ACID |
| Phenylalanine | F | PHE | PHENYLALANINE |
| Glycine | G | GLY | GLYCINE |
| Histidine | H | HIS | HISTIDINE |
| Isoleucine | I | ILE | ISOLEUCINE |
| Lysine | K | LYS | LYSINE |
| Leucine | L | LEU | LEUCINE |
| Methionine | M | MET | METHIONINE |
| Asparagine | N | ASN | ASPARAGINE |
| Proline | P | PRO | PROLINE |
| Glutamine | Q | GLN | GLUTAMINE |
| Arginine | R | ARG | ARGININE |
| Serine | S | SER | SERINE |
| Threonine | T | THR | THREONINE |
| Valine | V | VAL | VALINE |
| Tryptophan | W | TRP | TRYPTOPHAN |
| Tyrosine | Y | TYR | TYROSINE |
[Execution from Interface]
Add [PDB_MUTATE] keywords to the dialog that appears when you click .
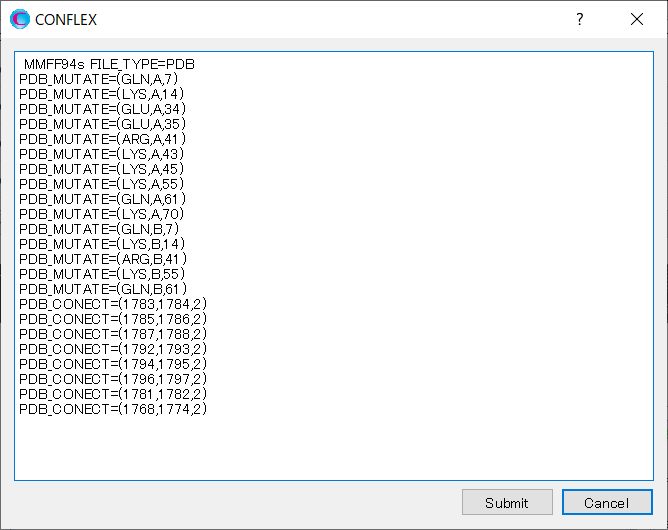 When you complete the modification, click to start the calculation.
When you complete the modification, click to start the calculation.
[Execution from command line]
Add [PDB_MUTATE] keywords to the 1ohr.ini file
1ohr.ini file
MMFF94S PDB_MUTATE=(GLN,A,7) PDB_MUTATE=(LYS,A,14) PDB_MUTATE=(GLU,A,34) PDB_MUTATE=(GLU,A,35) PDB_MUTATE=(ARG,A,41) PDB_MUTATE=(LYS,A,43) PDB_MUTATE=(LYS,A,45) PDB_MUTATE=(LYS,A,55) PDB_MUTATE=(GLN,A,61) PDB_MUTATE=(LYS,A,70) PDB_MUTATE=(GLN,B,7) PDB_MUTATE=(LYS,B,14) PDB_MUTATE=(ARG,B,41) PDB_MUTATE=(LYS,B,55) PDB_MUTATE=(GLN,B,61) PDB_CONECT=(1768,1774,2) PDB_CONECT=(1781,1782,2) PDB_CONECT=(1783,1784,2) PDB_CONECT=(1785,1786,2) PDB_CONECT=(1787,1788,2) PDB_CONECT=(1792,1793,2) PDB_CONECT=(1794,1795,2) PDB_CONECT=(1796,1797,2)
Store the two files of 1ohr.pdb and 1ohr.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par 1ohrenter
The above command is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
When you check the bso file after the caluclation has finished, you will get the following messages.
Information about "MUTATE RESIDUE" in the bso file
PDB_EXT: MUTATE RESIDUE FROM 7GLN TO GLN PDB_EXT: MUTATE RESIDUE FROM 14LYS TO LYS PDB_EXT: MUTATE RESIDUE FROM 34GLU TO GLU PDB_EXT: MUTATE RESIDUE FROM 35GLU TO GLU PDB_EXT: MUTATE RESIDUE FROM 41ARG TO ARG PDB_EXT: MUTATE RESIDUE FROM 43LYS TO LYS PDB_EXT: MUTATE RESIDUE FROM 45LYS TO LYS PDB_EXT: MUTATE RESIDUE FROM 55LYS TO LYS PDB_EXT: MUTATE RESIDUE FROM 61GLN TO GLN PDB_EXT: MUTATE RESIDUE FROM 70LYS TO LYS PDB_EXT: MUTATE RESIDUE FROM 7GLN TO GLN PDB_EXT: MUTATE RESIDUE FROM 14LYS TO LYS PDB_EXT: MUTATE RESIDUE FROM 41ARG TO ARG PDB_EXT: MUTATE RESIDUE FROM 55LYS TO LYS PDB_EXT: MUTATE RESIDUE FROM 61GLN TO GLN
To correct the missing atoms, you should specify the name of the original amino acid using the PDB_MUTATE keyword. If you want to change the amino acid sequence in the protein, you should specify a different amino acid name from the original using the PDB_MUTATE keyword.