Optimization and conformation search using Gaussian program
Geometry optimization and conformation search performed by CONFLEX are typically carried out using a classical molecular force field built into the software. While force field calculations are useful for quickly and efficiently locating energy minima, they cannot be performed if the required atom types and/or force field parameters for the target molecule are missing. In addition, it is possible that the distribution of conformational isomers obtained from a conformation search using a classical force field followed by re-optimization using quantum chemical methods may differ from that obtained using quantum chemical methods alone.
Starting from CONFLEX 8, Gaussian (16 or 09) can be invoked as an external program to perform geometry optimizations and conformation searches. This section outlines the settings required to use this functionality and presents the results of conformation searches conducted using both CONFLEX and Gaussian.
[Environment setting]
To use the Gaussian program in CONFLEX, you must also install Gaussian 16 or Gaussian 09 on your computer and set the appropriate environment variables to execute the Gaussian program.
In the cases of Mac and Linux
The environment variable g16root or g09root should be set. If the GAUSS_SCRDIR environment variable is not set, scratch files will be written to the same directory as the input files.
In the case of Windows
The environment variable GAUSS_EXEDIR should be set to the path of the folder where Gaussian is installed (e.g., C:¥G16W). If the [EXT_GAU_SCRDIR=] CONFLEX keyword is not used, scratch files will be written to %GAUSS_EXEDIR%¥Scratch.
By preparing a file with the necessary environment settings, you can enable the environment variables for the Gaussian program only during calculations.
[Conformation search of propylene glycol using B3LYP/6-31G(d) calculation]
This section explains how to perform a conformation search of propylene glycol using the B3LYP/6-31G(d) calculation as an example.
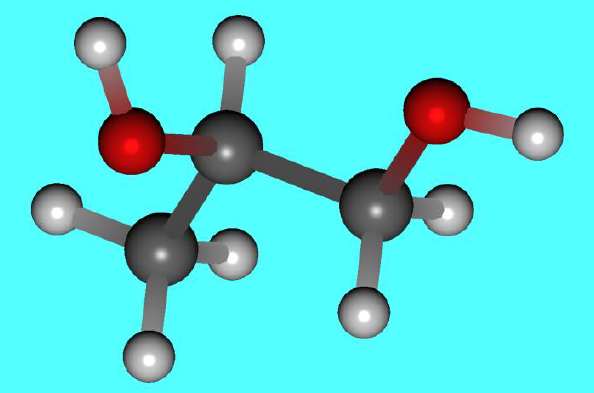 Input structure of propylene glycol
Input structure of propylene glycol
Structure data of propylene glycol (PropyleneGlycol.mol)
PropyleneGlycol.mol 13 12 0 0 0 0 0 0 0 0 0 0 -4.0461 -0.3459 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -3.6894 0.1585 0.8737 H 0 0 0 0 0 0 0 0 0 0 0 0 -3.6894 0.1585 -0.8737 H 0 0 0 0 0 0 0 0 0 0 0 0 -5.1161 -0.3459 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0 -3.5328 -1.7978 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -3.8895 -2.3022 0.8737 H 0 0 0 0 0 0 0 0 0 0 0 0 -1.9928 -1.7979 -0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -1.6361 -1.2935 -0.8737 H 0 0 0 0 0 0 0 0 0 0 0 0 -1.6361 -1.2934 0.8736 H 0 0 0 0 0 0 0 0 0 0 0 0 -4.0095 -2.4719 -1.1676 O 0 0 0 0 0 0 0 0 0 0 0 0 -4.5428 -3.2264 -0.9068 H 0 0 0 0 0 0 0 0 0 0 0 0 -1.5162 -3.1461 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0 -0.5757 -3.1535 0.1927 H 0 0 0 0 0 0 0 0 0 0 0 0 1 2 1 0 0 0 0 1 3 1 0 0 0 0 1 4 1 0 0 0 0 1 5 1 0 0 0 0 5 6 1 0 0 0 0 5 7 1 0 0 0 0 5 10 1 0 0 0 0 7 8 1 0 0 0 0 7 9 1 0 0 0 0 7 12 1 0 0 0 0 10 11 1 0 0 0 0 12 13 1 0 0 0 0 M END
[Execution from Interface]
Open the PropyleneGlycol.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
First, in [General Settings] dialog of the detailed settings dialog, select [Conformation Search] from the [Calculation Type:] pull-down menu.
Furthermore, select [External Program] from the pull-down menu of [Estimating Method:]. With this setting, geometry optimizations and others will be performed using the external program (Gaussian).
Next, configure the settings for the Gaussian program in the [External Program] dialog.
Set [Job Title:] as a comment to “Propylene Glycol B3LYP/6-31G(d) optimization”.
Set [%Mem=] to “128” and select [MW] from the pull-down menu for the unit.
Set [%NProcShared=] to “2”.
Set [Route Section:] to “B3LYP/6-31G(d) Opt” to perform geometry optimization using the B3LYP/6-31G(d) method.
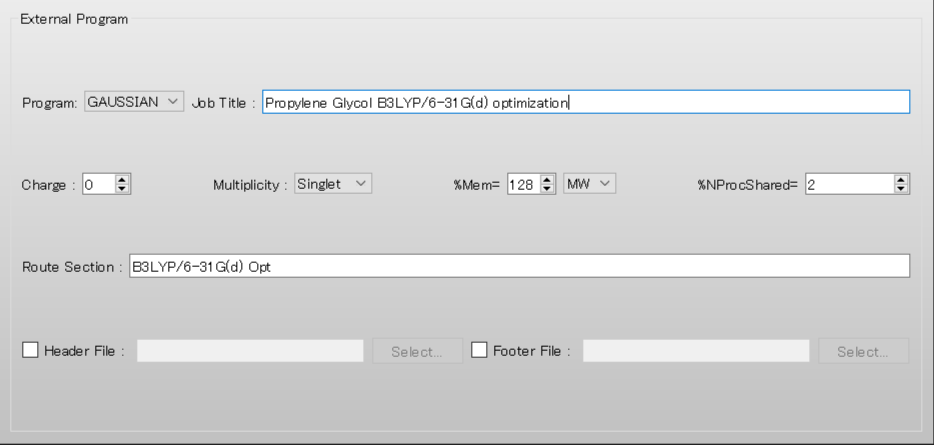
You can also configure the calculation settings for the Gaussian program using a file. For details, please refer to [Calculation setting of Gaussian program using a file].
Next, in [Conformation Search] dialog, set [Search Limit:] to “5.0”.
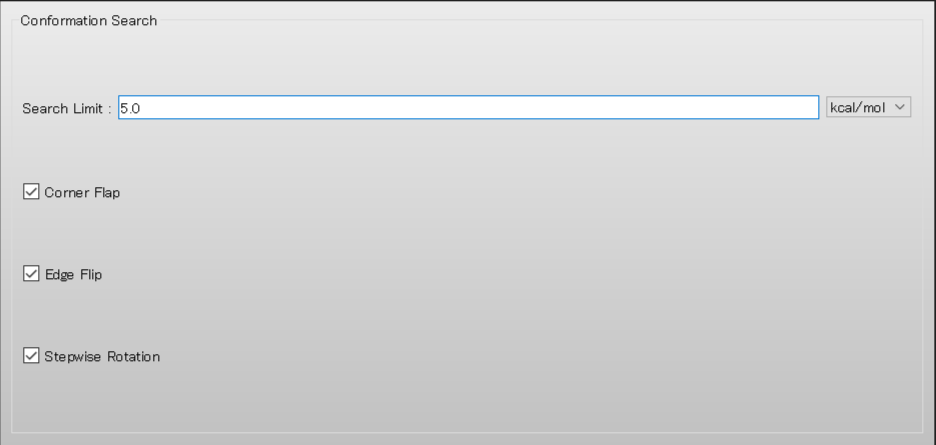 When completing the calculation settings, click in the detailed settings dialog.
When completing the calculation settings, click in the detailed settings dialog.
Add “EDIF_HARD=1.0D-3” keyword to the dialog that appears.
This sets the energy difference threshold to 0.001 kcal/mol. It is used to determine whether newly obtained conformations should be retained.
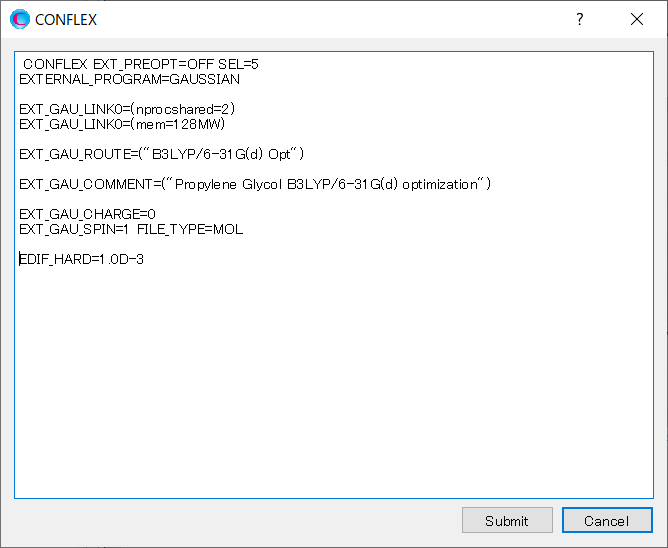 When completing the addition, click to start the calculation.
When completing the addition, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the PropyleneGlycol.ini file.
PropyleneGlycol.ini file
CONFLEX
SEL=5
EXTERNAL_PROGRAM=GAUSSIAN
EXT_GAU_LINK0=(mem=128MW)
EXT_GAU_LINK0=(nprocshared=2)
EXT_GAU_ROUTE=("B3LYP/6-31G(d) opt”)
EXT_GAU_COMMENT=("Propylene Glycol B3LYP/6-31G(d) optimization”)
EXT_GAU_CHARGE=0
EXT_GAU_SPIN=1
EXT_PREOPT=OFF
EDIF_HARD=1.0D-3
[CONFLEX] indicates that a conformation search will be performed.
[SEL=5] sets the search limit to 5 kcal/mol.
[EXTERNAL_PROGRAM=] specifies the name of the external program to be used.
[EXT_GAU_LINK0=] defines a Link0 command (this keyword can be used multiple times in the INI file).
[EXT_GAU_ROUTE=] specifies the route section.
[EXT_GAU_COMMENT=] specifies a comment.
[EXT_GAU_CHARGE=] sets the total charge of the system.
[EXT_GAU_SPIN=] sets the spin multiplicity of the system.
[EXT_PREOPT=OFF] means that geometry optimization using a force field will not be performed before the Gaussian calculation.
[EDIF_HARD=1.0D-3] sets the energy difference threshold to 0.001 kcal/mol. This threshold is used to determine whether newly generated conformations should be retained.
Note that both [EXT_GAU_ROUTE=] and [EXT_GAU_COMMENT=] must be enclosed in (“] and [”).
You can also configure the calculation settings for the Gaussian program using a file. For details, please refer to [Calculation setting of Gaussian program using a file].
Store the two files of PropyleneGlycol.mol and PropyleneGlycol.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par PropyleneGlycolenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
A portion of the output in the PropyleneGlycol.ls1 file is shown below. The input/output file names for Gaussian correspond to the [CONF. ID] listed in the .ls1 file. The files PropyleneGlycol_00006.log and PropyleneGlycol_00006.fchk contain the data for the most stable structure. The [ORIGINAL STERIC E] field in the .ls1 file shows the energy obtained from the Gaussian calculation, converted to kcal/mol. Note that Gaussian uses atomic units (a.u.). The total energy of the most stable structure is -269.56133526 a.u., which corresponds to -169,152.2987 kcal/mol. In this tutorial, we used Gaussian 16 Rev. A.03.
PropyleneGlycol.ls1:
====================================================================================
CONF. ORIGINAL DISTRI- NO.
NO. ID STERIC E DELTA E BUTION INIT. REOPT. NEG.
------------------------------------------------------------------------------------
1 00000006 -169152.2987 0.0000 21.2884 T* F+ 0
2 00000017 -169152.2709 0.0278 20.3129 T* F+ 0
3 00000019 -169152.1497 0.1490 16.5548 T* F+ 0
4 00000011 -169151.9408 0.3579 11.6356 T* F+ 0
5 00000001 -169151.9312 0.3675 11.4487 T* F+ 0
6 00000008 -169151.6798 0.6189 7.4899 T* F+ 0
7 00000012 -169151.4409 0.8578 5.0044 T* F+ 0
8 00000018 -169151.1376 1.1611 2.9993 T* F+ 0
9 00000007 -169150.9622 1.3365 2.2310 T* F+ 0
10 00000016 -169149.5319 2.7668 0.1995 T* F+ 0
11 00000020 -169149.3686 2.9301 0.1515 T* F+ 0
12 00000009 -169149.2519 3.0468 0.1244 T* F+ 0
13 00000022 -169149.2130 3.0857 0.1165 T* F+ 0
14 00000021 -169149.0687 3.2300 0.0913 T* F+ 0
15 00000023 -169149.0383 3.2604 0.0868 T* F+ 0
16 00000024 -169149.0232 3.2755 0.0846 T* F+ 0
17 00000013 -169148.9887 3.3101 0.0798 T* F+ 0
18 00000003 -169148.8937 3.4050 0.0680 T* F+ 0
19 00000010 -169147.6211 4.6776 0.0079 T* F+ 0
20 00000014 -169147.4593 4.8394 0.0060 T* F+ 0
21 00000004 -169147.3564 4.9423 0.0051 T* F+ 0
22 00000015 -169147.3371 4.9616 0.0049 T* F+ 0
23 00000005 -169147.3181 4.9806 0.0048 T* F+ 0
24 00000002 -169147.2083 5.0904 0.0040 T* F+ 0
------------------------------------------------------------------------------------
MINIMUM ENERGY: -169152.2987 KCAL/MOL
AVERAGE ENERGY: -169151.9983 KCAL/MOL (BASED ON THE BOLTZMANN LAW)
====================================================================================
List of Gaussian input/output files:
PropyleneGlycol_00001.fchk PropyleneGlycol_00008.fchk PropyleneGlycol_00015.fchk PropyleneGlycol_00001.gjf PropyleneGlycol_00008.gjf PropyleneGlycol_00015.gjf PropyleneGlycol_00001.log PropyleneGlycol_00008.log PropyleneGlycol_00015.log PropyleneGlycol_00002.fchk PropyleneGlycol_00009.fchk PropyleneGlycol_00016.fchk PropyleneGlycol_00002.gjf PropyleneGlycol_00009.gjf PropyleneGlycol_00016.gjf PropyleneGlycol_00002.log PropyleneGlycol_00009.log PropyleneGlycol_00016.log PropyleneGlycol_00003.fchk PropyleneGlycol_00010.fchk PropyleneGlycol_00017.fchk ・・・・・
When the conformation search of propylene glycol is performed using the MMFF94s force field with a search limit of 7 kcal/mol, 21 conformers are obtained.
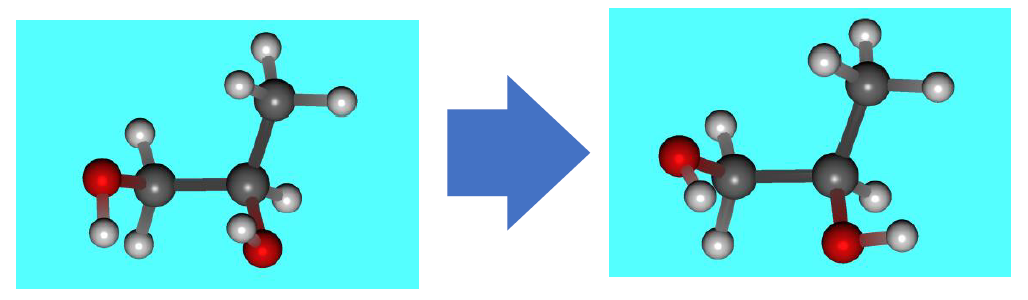
After reoptimization at the B3LYP/6-31G(d) level, the 5th conformer from the MMFF94s search becomes the most stable conformation (as shown in the figure below).
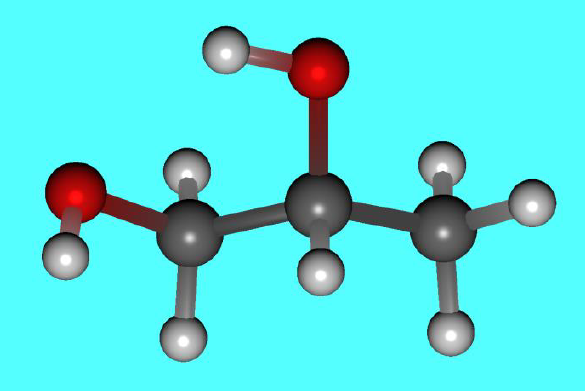 The most stable structure of propylene glycol at B3LYP/6-31G(d) level
The most stable structure of propylene glycol at B3LYP/6-31G(d) level
Furthermore, the 10th conformer from the MMFF94s search, when re-optimized by Gaussian, converges to a different conformation (see the figure below). As the result, the number of distinct conformers at the B3LYP/6-31G(d) level is 20.
When comparing the conformations obtained using only the B3LYP/6-31G(d) calculation with those obtained by reoptimizing the conformers generated by the MMFF94s calculation, it can be seen that the former yields 4 new stable conformers (No. 19, 20, 23, and 24).
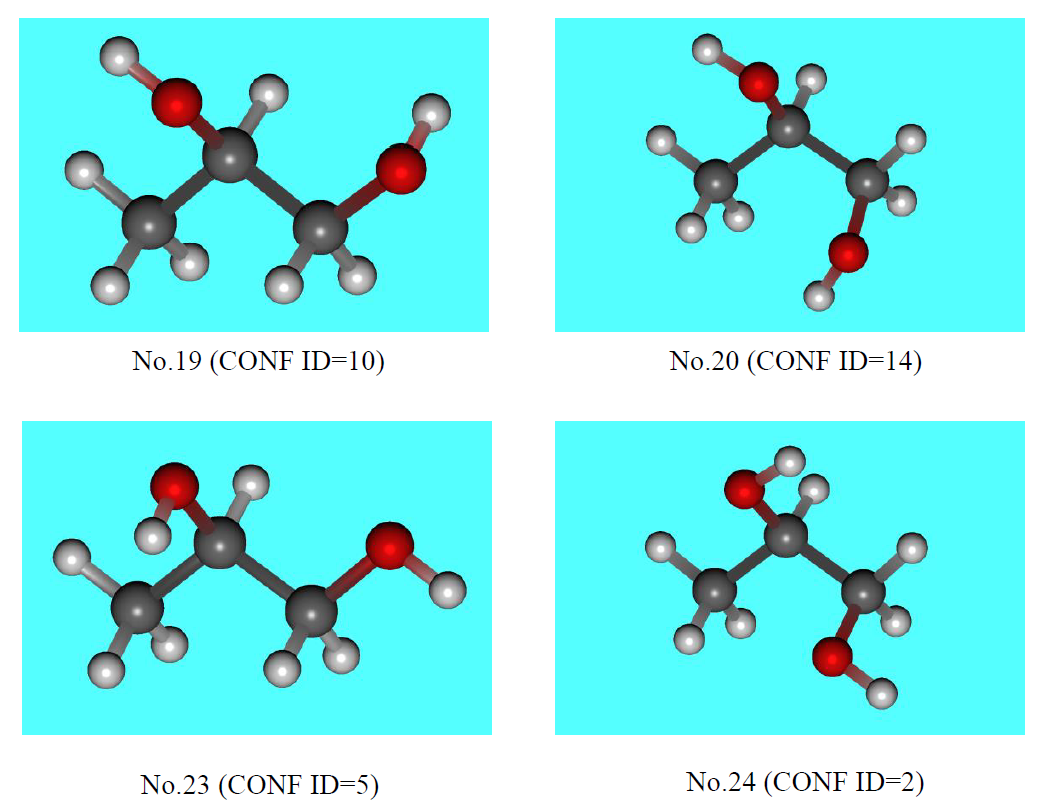 Conformers obtained only from the conformation search at B3LYP/6-31G(d) level
Conformers obtained only from the conformation search at B3LYP/6-31G(d) level
[Calculation setting of Gaussian program using a file]
We can configure the Link0 command and route section using a file.
* Example 1
An input file for the Gaussian program is shown below.
%mem=32MW %nprocshared=2 #HF/3-21G opt test of HF/3-21G calculation 0 1 molecular specification ...
Save the settings above the molecular specification as a gaussian.hdr file.
gaussian.hdr file:
%mem=32MW %nprocshared=2 #HF/3-21G opt test of HF/3-21G calculation 0 1
[Execution from Interface]
In [External Program] dialog of the detailed settings dialog, check the [Header File:] checkbox.
Next, click [Select...], and select the gaussian.hdr file. Gaussian settings are automatically configured according to the gaussian.hdr file.
[Execution from command line]
Modify the PropyleneGlycol.ini file as shown below.
PropyleneGlycol.ini file
CONFLEX
SEL=5
EXTERNAL_PROGRAM=GAUSSIAN
EXT_GAU_HEADER_FILE=(gaussian.hdr)
EXT_PREOPT=OFF
EDIF_HARD=1.0D-3
Store the three files of PropyleneGlycol.mol, PropyleneGlycol.ini and gaussian.hdr in a single folder, and execute CONFLEX.
* Example 2
An input file for the Gaussian program is shown below. The settings below the molecular specification, such as the basis set settings defined by the Gen keyword, can also be specified using a file.
%mem=20MW %nprocshared=2 # B3LYP/gen Opt TEST OF B3LYP/6-31G(d) CALCULATION 0 1 molecular specification ... H C O 0 6-31G(d) ****
Save the settings above the molecular specification as a gaussian.hdr file.
gaussian.hdr file:
%mem=32MW %nprocshared=2 #HF/3-21G opt test of HF/3-21G calculation 0 1
Save the settings below the molecular specification as a gaussian.ftr file.
gaussian.ftr file:
H C O 0 6-31G(d) ****
[Execution from Interface]
In [External Program] dialog of the detailed settings dialog, check the [Header File:] checkbox.
Next, click [Select...] and select the gaussian.hdr file.
Also check the [Footer File:] checkbox.
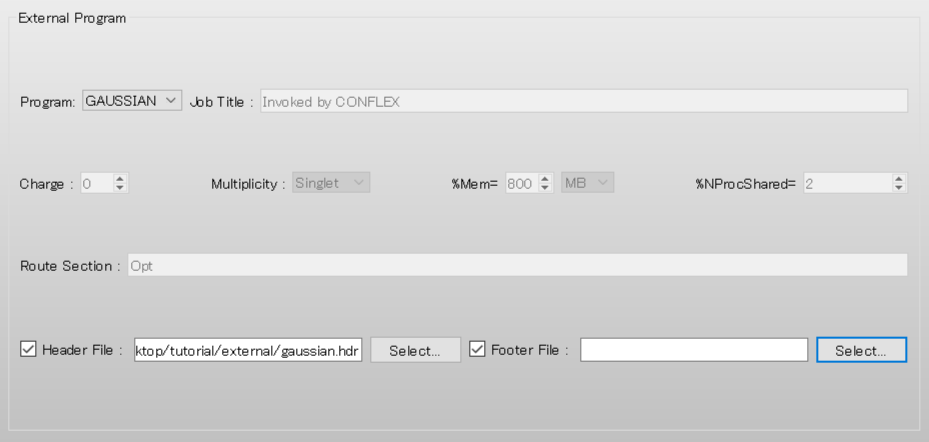 Next, click [Select...] and select the gaussian.ftr file.
Next, click [Select...] and select the gaussian.ftr file.
Gaussian settings are automatically configured according to the gaussian.hdr and gaussian.ftr files.
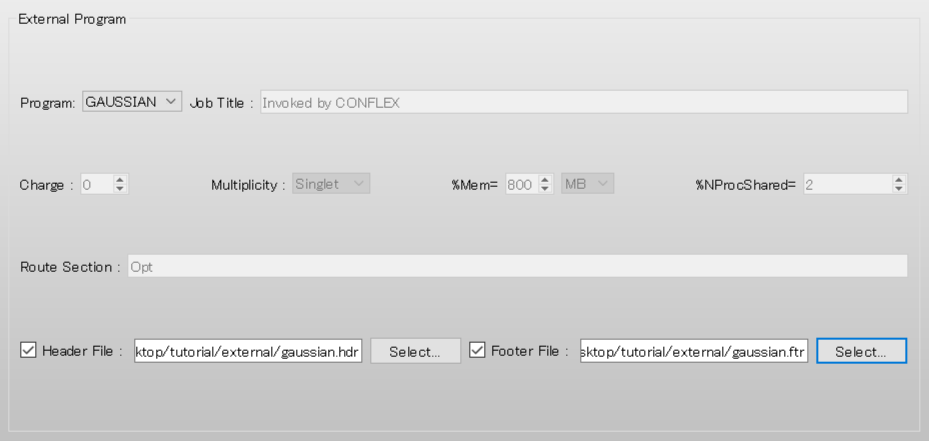
[Execution from command line]
Modify the PropyleneGlycol.ini file as shown below.
PropyleneGlycol.ini file
CONFLEX SEL=5 EXTERNAL_PROGRAM=GAUSSIAN EXT_GAU_HEADER_FILE=(gaussian.hdr) EXT_GAU_FOOTER_FILE=(gaussian.ftr) EXT_PREOPT=OFF EDIF_HARD=1.0D-3
Store the four files of PropyleneGlycol.mol, PropyleneGlycol.ini, gaussian.hdr, and gaussian.ftr in a single folder, and execute CONFLEX.
[Execution of Gaussian using a job scheduler (Linux)]
This section explains how to execute Gaussian jobs created by CONFLEX using a job scheduler. In this case, we use the Univa Grid Engine job scheduler.
[Execution from command line]
Modify the PropyleneGlycol.ini file as shown below.
PropyleneGlycol.ini file
CONFLEX SEL=5 EXTERNAL_PROGRAM=GAUSSIAN EXT_JOB_COM=("qsub -sync yes”) EXT_JOB_FILE=qsub.sh EXT_GAU_LINK0=(mem=128MW) EXT_GAU_LINK0=(nprocshared=2) EXT_GAU_ROUTE=("B3LYP/6-31G(d) opt”) EXT_GAU_COMMENT=("Propylene Glycol B3LYP/6-31G(d) optimization”) EXT_GAU_CHARGE=0 EXT_GAU_SPIN=1 EXT_PREOPT=OFF EDIF_HARD=1.0D-3
[EXT_JOB_COM=] specifies the execution command for the job scheduler. When using the job scheduler, you should include the option "-sync yes" to submit the job interactively.
[EXT_JOB_FILE=] specifies a file that contains the settings for the job scheduler.
qsub.sh file:
#!/bin/bash #$ -S /bin/bash #$ -N g16_from_conflex #$ -cwd #$ -V #$ -j y #$ -q all.q #$ -pe sme 2 export g16root=/usr/local
Gaussian jobs will be executed with the command [qsub -sync yes qsub.sh]. The qsub.sh file does not contain the Gaussian execution command, but CONFLEX adds it automatically. You should store the qsub.sh file in the same folder that contains the PropyleneGlycol.mol and PropyleneGlycol.ini files.
[Temporarily enable Gaussian preferences]
You can enable the environment settings for the Gaussian program only when performing Gaussian calculations.
In the case of Windows
[Execution from Interface]
Click on the detailed settings dialog, and add [EXT_JOB_FILE=setup.txt] to the dialog that appears.
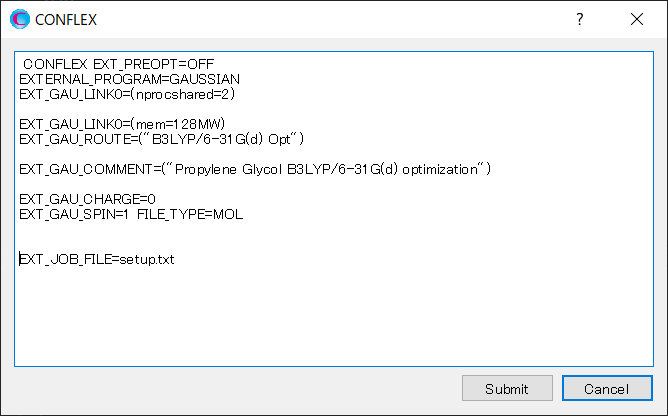
[Execution from command line]
Modify the PropyleneGlycol.ini file as shown below.
PropyleneGlycol.ini file
CONFLEX
SEL=5
EXTERNAL_PROGRAM=GAUSSIAN
EXT_JOB_FILE=setup.txt
EXT_GAU_LINK0=(mem=128MW)
EXT_GAU_LINK0=(nprocshared=2)
EXT_GAU_ROUTE=("B3LYP/6-31G(d) opt”)
EXT_GAU_COMMENT=("Propylene Glycol B3LYP/6-31G(d) optimization”)
EXT_GAU_CHARGE=0
EXT_GAU_SPIN=1
EXT_PREOPT=OFF
EDIF_HARD=1.0D-3
[EXT_JOB_FILE=] specifies the name of the file for setting the environment variables. When you use this keyword, CONFLEX creates a bat file containing the contents of the setup.txt file and the Gaussian execution command, then executes the job using the batch file.
setup.txt file:
set GAUSS_EXEDIR=C:\G16W
In addition, add [EXT_GAU_SCRDIR=D:\Scratch] to the dialog that appears by clicking . This allows you to save scratch files to the specified folder. If you don't add this, the scratch files will be saved in [%GAUSS_EXEDIR%¥Scratch].
In the cases of Mac and Linux
[Execution from Interface]
Click on the detailed settings dialog, and add [EXT_JOB_COM=(“sh")] and [EXT_JOB_FILE=setup.sh] to the dialog that appears.
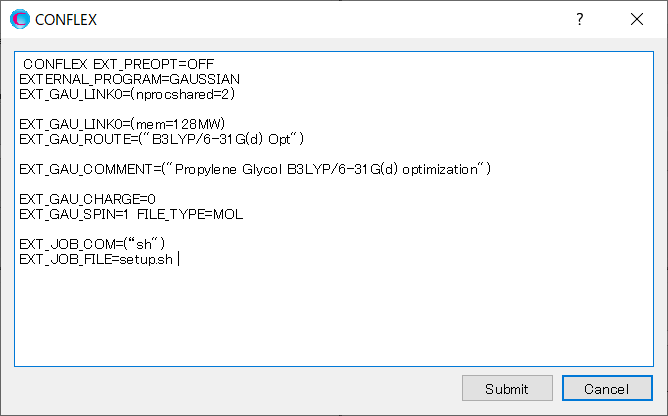
[Execution from command line]
Modify the PropyleneGlycol.ini file as shown below.
PropyleneGlycol.ini file
CONFLEX SEL=5 EXTERNAL_PROGRAM=GAUSSIAN EXT_JOB_COM=("sh”) EXT_JOB_FILE=setup.sh EXT_GAU_LINK0=(mem=128MW) EXT_GAU_LINK0=(nprocshared=2) EXT_GAU_ROUTE=("B3LYP/6-31G(d) opt”) EXT_GAU_COMMENT=("Propylene Glycol B3LYP/6-31G(d) optimization”) EXT_GAU_CHARGE=0 EXT_GAU_SPIN=1 EXT_PREOPT=OFF EDIF_HARD=1.0D-3
[EXT_JOB_COM=] specifies the execution command, and [EXT_JOB_FILE=] specifies the name of the file for setting the environment variables. When you use these keywords, CONFLEX creates a script file containing the contents of the setup.sh file and the Gaussian execution command, and executes the job using the sh command and the script file.
setup.sh file:
export g16root=/usr/local export GAUSS_SCRDIR=/tmp