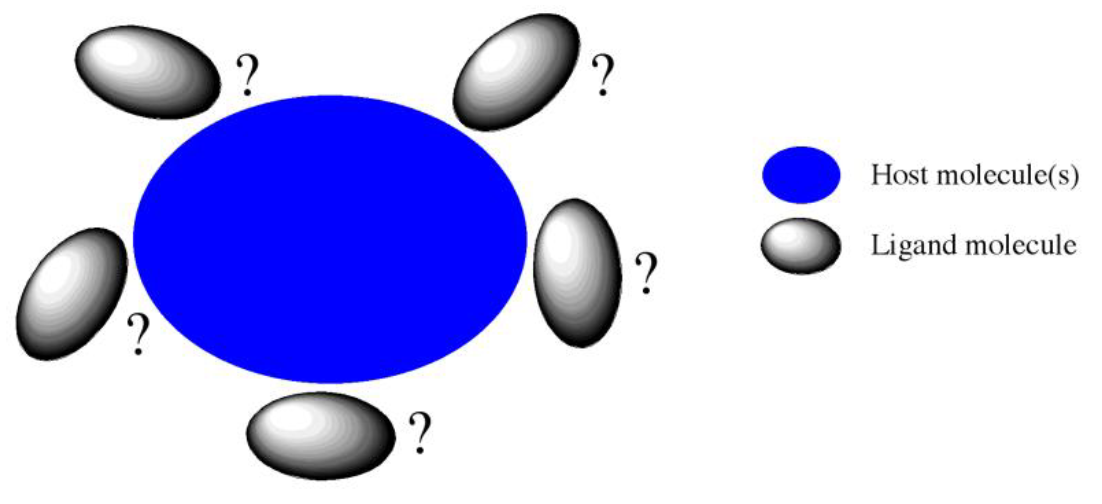
Host-Ligand coordination search
CONFLEX has a function called “Host-Ligand coordination search”. It is useful for finding stable structures of complex molecules or molecular clusters by exploring stable relative positions and orientations of a molecule with respect to other molecule(s). In this search method, the molecule(s) are defined as “host”, and the other molecule or ionic species is defined as the “ligand”. The ligand is automatically arranged around the host (refer to Figure 1). This function is expected to be applied in supramolecular chemistry, such as molecular recognition or host-guest chemistry.
[Overview of search method]
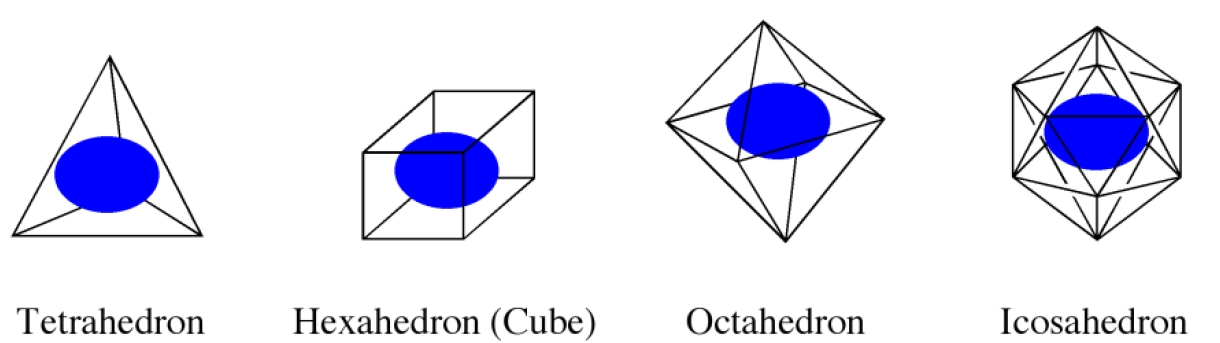
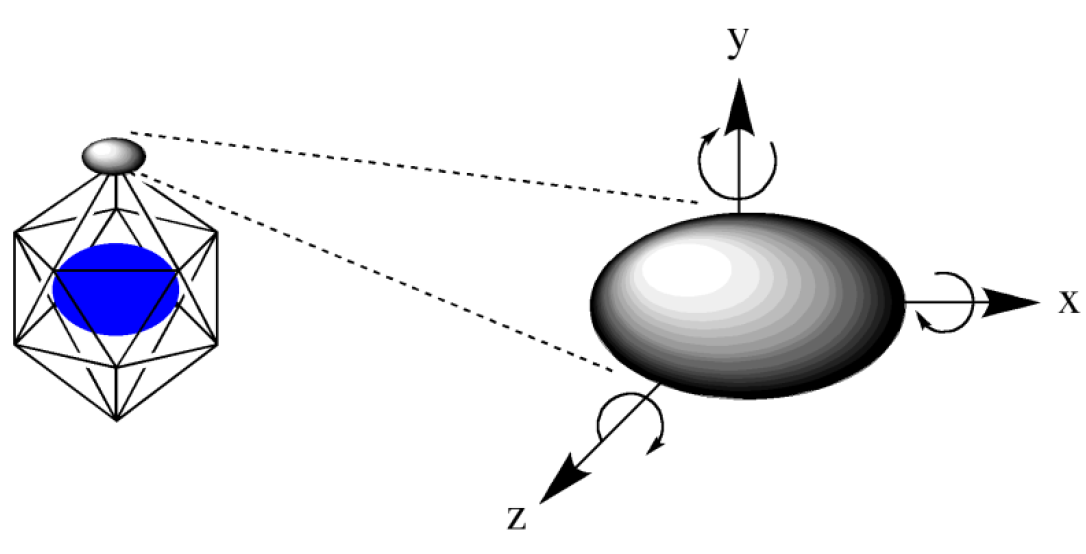
First, we choose a regular polyhedron to surround the host molecule(s). CONFLEX supports four polyhedron models: tetrahedron, hexahedron, octahedron, and icosahedron (Figure 2). The icosahedron is used as the default setting.
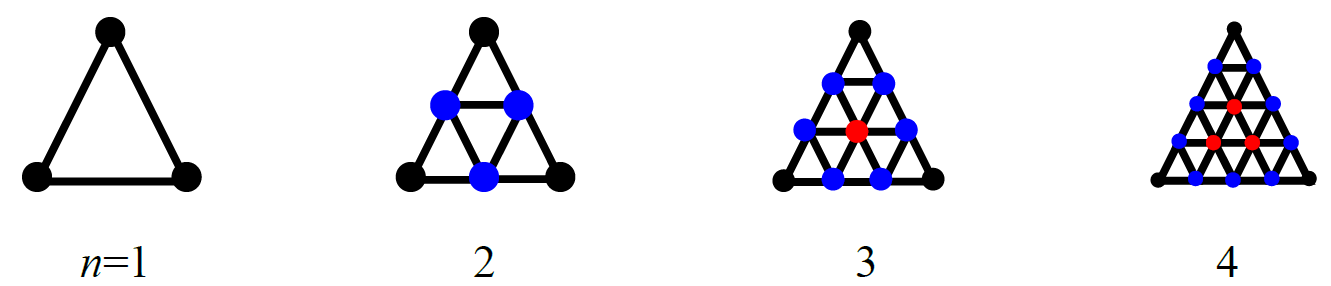
CONFLEX arranges the ligand molecule on the vertices of the regular polyhedron. If you want to explore stable structures in more detail, you can increase the number of points for arranging the ligand by dividing each face of the regular polyhedron using the “HLSEARCH_HOST_NDIV=n” keyword (Figure 3).
The arranged ligand is rotated around the x, y, and z axes to generate initial coordination structures (Figure 4). The initial structures are then subjected to geometry optimization, and the optimized structures are output in order of energy. The rotation angles can be specified using the keyword “HLSEARCH_LIGAND_ROT=(l,m,n)” with the default setting being 60 degrees (l=m=n=6). When the keyword “HLSEARCH_LIGAND_ROT=(8,8,8)” is used, the rotation angles are set to 45 degrees, respectively.
[Stable coordinations of two acetic acid molecules]
This section applies the Host-Ligand coordination search to a molecular complex of two acetic acid molecules.
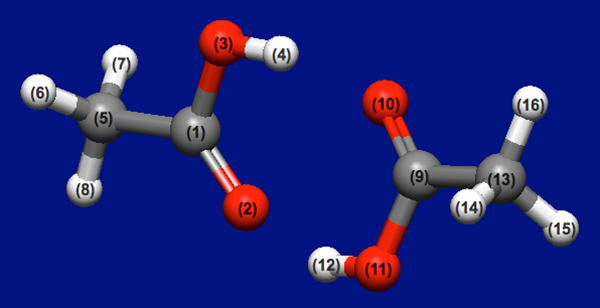
Structure data (acetic_acid_dimer.mol)
acetic_acid_dimer.mol
16 14 0 0 0 0 0 0 0 0 0 0
0.9628 -0.5511 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1931 -0.5511 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.3335 0.6511 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.9803 1.3755 -0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.0286 -1.7296 0.0001 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.6441 -1.6778 0.8738 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.6442 -1.6778 -0.8735 H 0 0 0 0 0 0 0 0 0 0 0 0
0.5138 -2.6519 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
3.5500 2.4393 0.3355 C 0 0 0 0 0 0 0 0 0 0 0 0
2.4107 2.2978 -0.1068 O 0 0 0 0 0 0 0 0 0 0 0 0
4.2100 1.3256 0.7422 O 0 0 0 0 0 0 0 0 0 0 0 0
3.6576 0.5365 0.6183 H 0 0 0 0 0 0 0 0 0 0 0 0
4.3924 3.7159 0.5151 C 0 0 0 0 0 0 0 0 0 0 0 0
4.6407 3.8369 1.5488 H 0 0 0 0 0 0 0 0 0 0 0 0
5.2907 3.6344 -0.0604 H 0 0 0 0 0 0 0 0 0 0 0 0
3.8309 4.5636 0.1817 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0 0 0 0
1 3 1 0 0 0 0
1 5 1 0 0 0 0
3 4 1 0 0 0 0
5 6 1 0 0 0 0
5 7 1 0 0 0 0
5 8 1 0 0 0 0
9 10 2 0 0 0 0
9 11 1 0 0 0 0
9 13 1 0 0 0 0
11 12 1 0 0 0 0
13 14 1 0 0 0 0
13 15 1 0 0 0 0
13 16 1 0 0 0 0
M END
[Execution from Interface]
Open the acetic_acid_dimer.mol file using CONFLEX Interface.
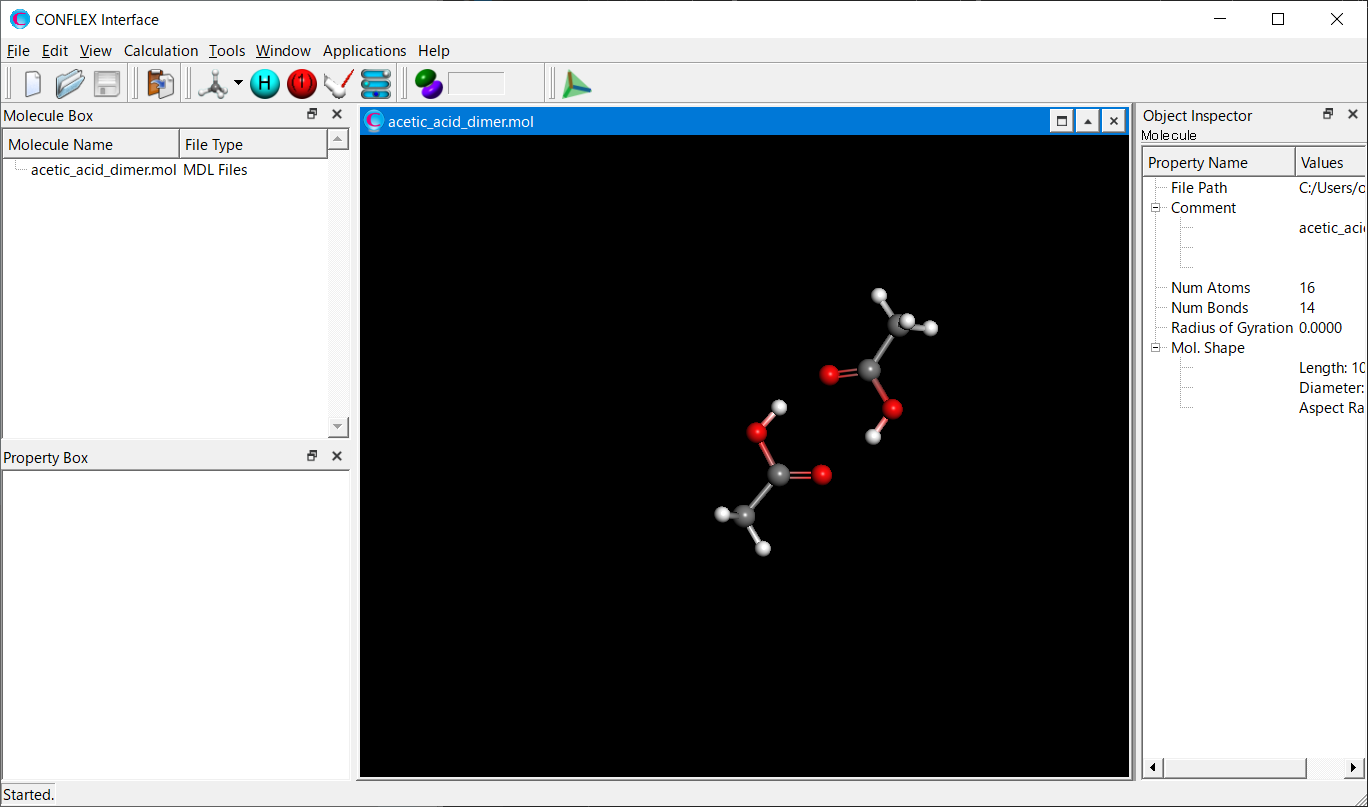
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears.
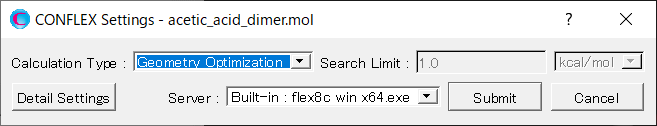
Next, in [General Settings] dialog of the detailed settings dialog, select [Host Ligand Search] from the pull-down menu of [Calculation Type:].
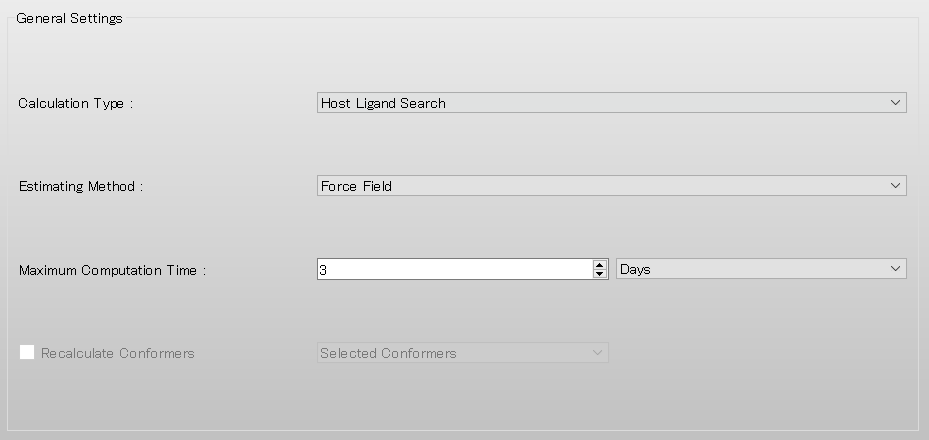
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the acetic_acid_dimer.ini file.
acetic_acid_dimer.ini file
MMFF94S HLSEARCH
[HLSEARCH] indicates that a Host-Ligand coordination search is to be performed.
[MMFF94s] means to use MMFF94s force field.
Store the two files of acetic_acid_dimer.mol and acetic_acid_dimer.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par acetic_acid_dimerenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
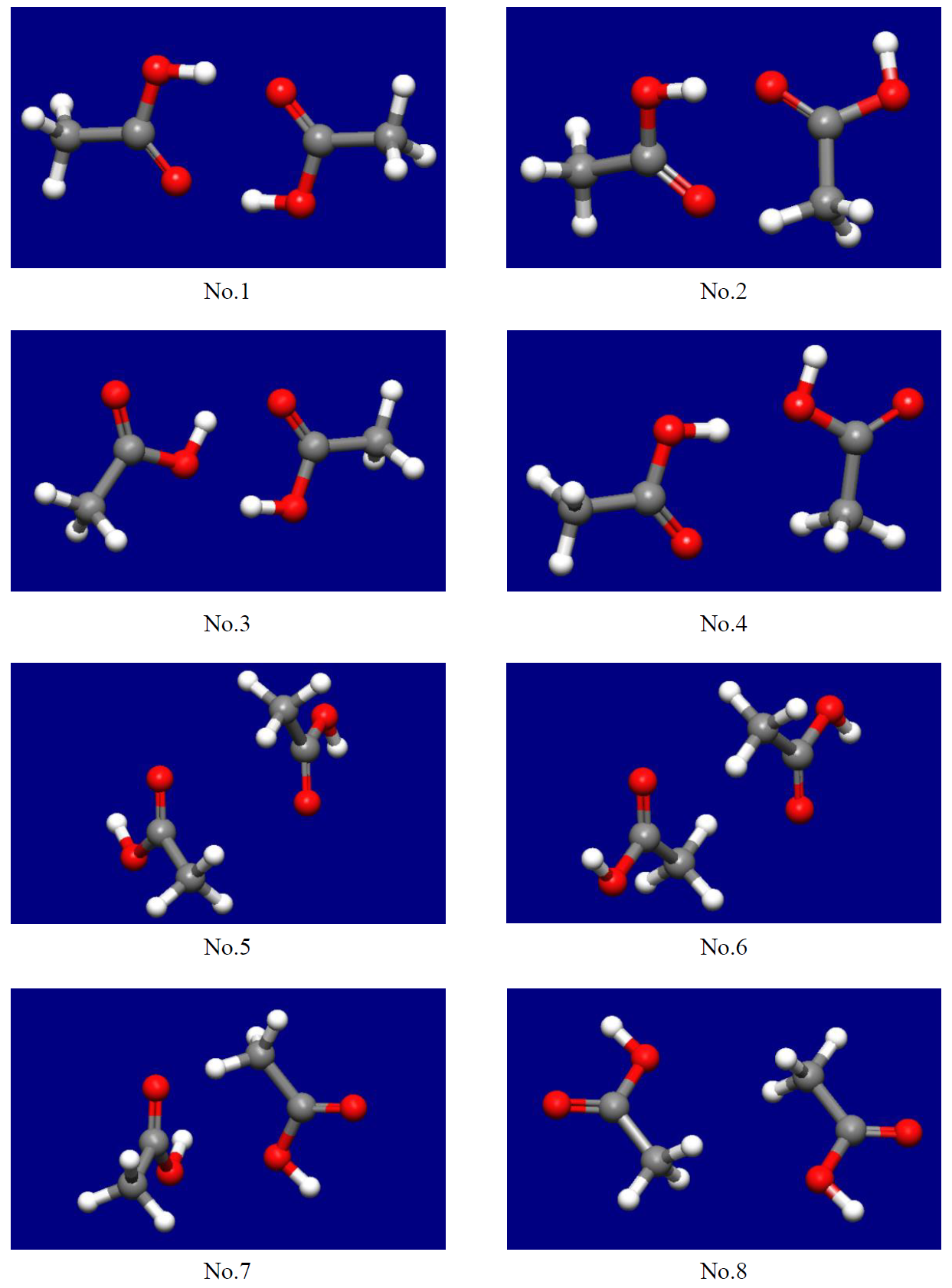
As the results, we obtain 8 stable structures of the molecular complex (see following figure).
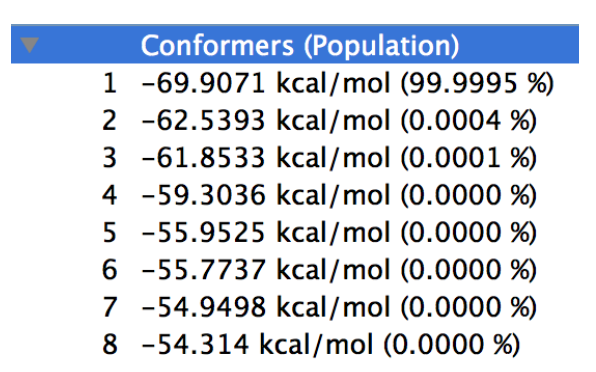
The energy list is displayed in the Property Box of CONFLEX Interface, as shown in the figure below.
About how to visualize the structures, please refer to [Visualization of calculation results].
[Coordination search of glucose and water molecules]
This section demonstrates the Host-Ligand coordination search applied to a molecular complex consisting of α-D-glucose and water molecules.
In this case, α-D-glucose is treated as the host molecule, and water is treated as the ligand.
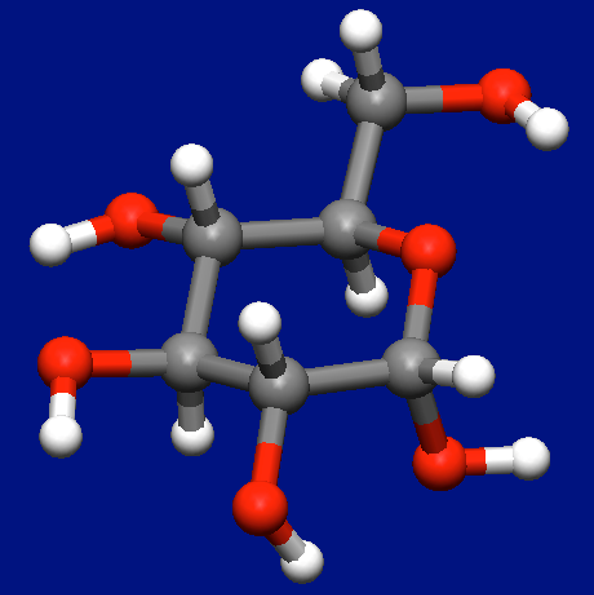
First, the most stable conformer of α-D-glucose (see figure below) is obtained through a conformation search using the MMFF94s force field.
Next, a molecular complex consisting of α-D-glucose and water molecules is created by adding a water molecule to the most stable conformer (see figure below).
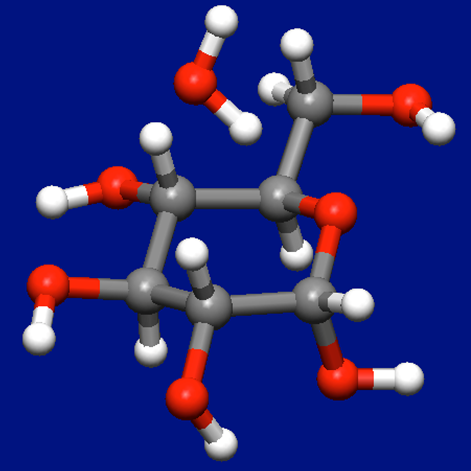
Structure data of the molecular complex for α-D-glucose and water molecules (aDglucose_H2O.mol)
aDglucose_H2O.mol
27 26 0 0 0 999 V2000
-0.5024 2.4148 0.6016 O 0 0 0 0 0
-0.8520 -1.0932 -0.5731 O 0 0 0 0 0
0.4519 -1.5744 -0.2774 C 0 0 0 0 0
-0.2020 1.2411 -0.1553 C 0 0 0 0 0
-1.1949 0.1125 0.1452 C 0 0 0 0 0
2.1431 1.8092 -0.2200 O 0 0 0 0 0
0.5330 -1.9764 1.0847 O 0 0 0 0 0
2.8319 -0.9820 -0.1321 O 0 0 0 0 0
-2.6186 0.5024 -0.2701 C 0 0 0 0 0
1.5391 -0.5233 -0.5592 C 0 0 0 0 0
1.2184 0.7799 0.1723 C 0 0 0 0 0
-3.5094 -0.5881 -0.0183 O 0 0 0 0 0
0.2794 2.9953 0.4978 H 0 0 0 0 0
0.6445 -2.4621 -0.8896 H 0 0 0 0 0
-0.2535 1.5201 -1.2154 H 0 0 0 0 0
-1.2141 -0.1177 1.2184 H 0 0 0 0 0
3.0267 1.3889 -0.1951 H 0 0 0 0 0
-0.0183 -2.7768 1.1469 H 0 0 0 0 0
2.6839 -1.4193 0.7318 H 0 0 0 0 0
-2.6702 0.7196 -1.3421 H 0 0 0 0 0
-2.9791 1.3731 0.2851 H 0 0 0 0 0
1.6027 -0.3262 -1.6358 H 0 0 0 0 0
1.3472 0.6668 1.2558 H 0 0 0 0 0
-3.0727 -1.3725 -0.4004 H 0 0 0 0 0
-0.3106 0.8234 -2.7522 O 0 0 0 0 0
-0.3290 0.0085 -2.2451 H 0 0 0 0 0
-0.9697 0.7822 -3.4490 H 0 0 0 0 0
1 4 1 0 0 0 0
1 13 1 0 0 0 0
2 3 1 0 0 0 0
2 5 1 0 0 0 0
3 7 1 0 0 0 0
3 10 1 0 0 0 0
3 14 1 0 0 0 0
4 5 1 0 0 0 0
4 11 1 0 0 0 0
4 15 1 0 0 0 0
5 9 1 0 0 0 0
5 16 1 0 0 0 0
6 11 1 0 0 0 0
6 17 1 0 0 0 0
7 18 1 0 0 0 0
8 10 1 0 0 0 0
8 19 1 0 0 0 0
9 12 1 0 0 0 0
9 20 1 0 0 0 0
9 21 1 0 0 0 0
10 11 1 0 0 0 0
10 22 1 0 0 0 0
11 23 1 0 0 0 0
12 24 1 0 0 0 0
25 26 1 0 0 0 0
25 27 1 0 0 0 0
M END
[Execution from Interface]
Open the aDglucose_H2O.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears.
Next, in [General Settings] dialog of the detailed settings dialog, select [Host Ligand Search] from the pull-down menu of [Calculation Type:]. After completing the setting, click on the detailed settings dialog.
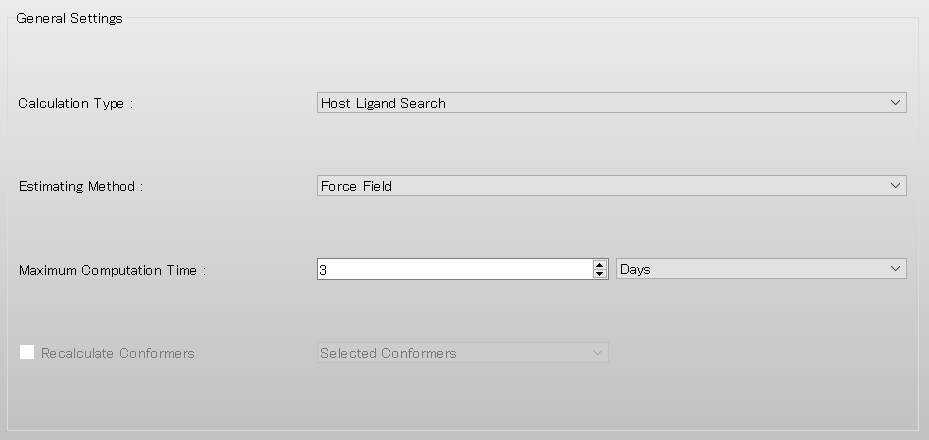
Add [OPT=GROUP] and [MOL_GROUP=(25,1)] to the dialog that appears.
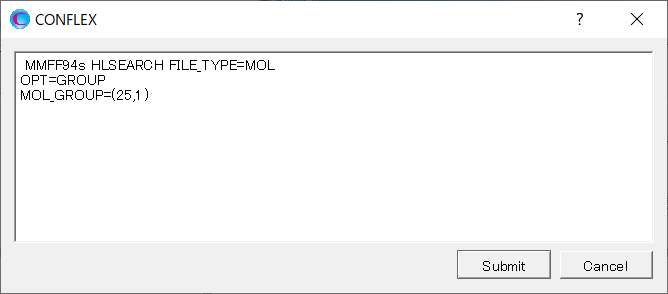
Here, the settings [OPT=GROUP] and [MOL_GROUP=(25,1)] indicate that the position and geometry of the water molecule are optimized, while those of α-D-glucose are fixed during the calculation.
For more details on geometry optimization with constraints, please refer to [Constraint by molecular object method].
After completing the modifications, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the aDglucose_H2O.ini file.
aDglucose_H2O.ini file
HLSEARCH OPT=GROUP MOL_GROUP=(25,1)
[HLSEARCH] indicates that a Host-Ligand coordination search is to be performed.
The settings [OPT=GROUP] and [MOL_GROUP=(25,1)] indicate that the position and geometry of the water molecule are optimized, while those of α-D-glucose are fixed during the calculation.
For more details on geometry optimization with constraints, please refer to [Constraint by molecular object method].
Store the two files of aDglucose_H2O.mol and aDglucose_H2O.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par aDglucose_H2Oenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
As the results, we obtaine 10 stable structures of the molecular complex of α-D-glucose and water molecules. The most stable structure is shown in the following figure.
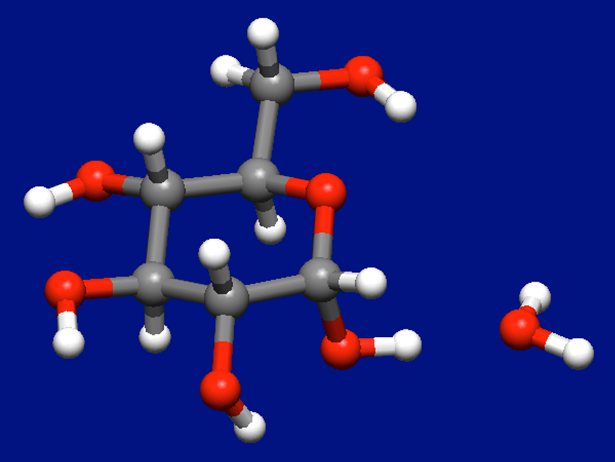
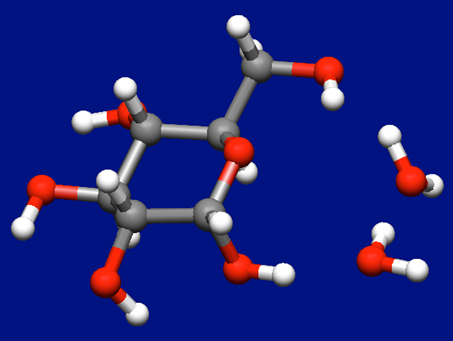
In case where two water molecules coordinate to the α-D-glucose
We add one more water molecule to the most stable structure of the α-D-glucose–water complex shown above, and perform the Host-Ligand coordination search again.
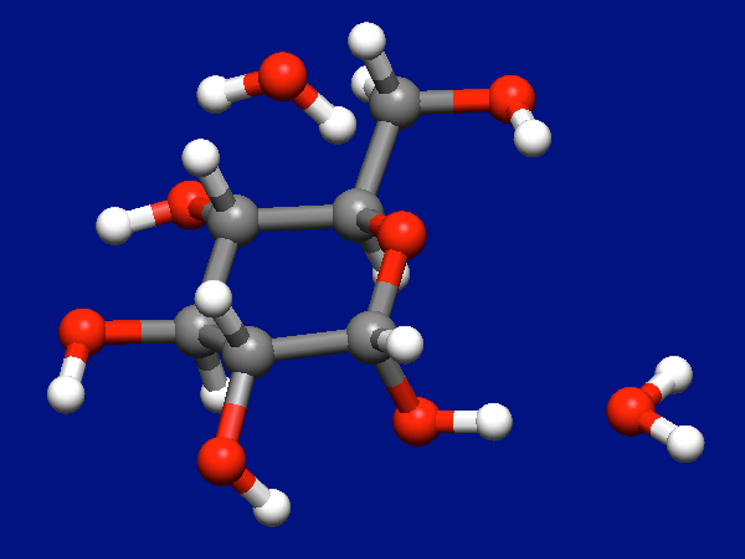
Structure data
aDglucose_2H2O.mol
30 28 0 0 999 V2000
-1.7903 2.0810 -0.8582 O 0 0 0 0 0
0.9497 0.1820 0.7911 O 0 0 0 0 0
0.4899 -1.1464 0.5531 C 0 0 0 0 0
-1.1899 1.1508 0.0453 C 0 0 0 0 0
0.3377 1.1585 -0.0836 C 0 0 0 0 0
-3.1447 -0.2633 -0.0402 O 0 0 0 0 0
0.9130 -1.5822 -0.7345 O 0 0 0 0 0
-1.4750 -2.5911 0.2513 O 0 0 0 0 0
0.9177 2.5280 0.2908 C 0 0 0 0 0
-1.0411 -1.2797 0.6520 C 0 0 0 0 0
-1.7220 -0.2507 -0.2493 C 0 0 0 0 0
2.3453 2.4922 0.1999 O 0 0 0 0 0
-2.7454 1.8632 -0.8498 H 0 0 0 0 0
0.9541 -1.8120 1.2891 H 0 0 0 0 0
-1.4901 1.4462 1.0585 H 0 0 0 0 0
0.6456 0.9385 -1.1142 H 0 0 0 0 0
-3.3916 -1.2089 0.0143 H 0 0 0 0 0
1.8682 -1.7501 -0.6492 H 0 0 0 0 0
-0.9383 -2.8164 -0.5360 H 0 0 0 0 0
0.6697 2.7883 1.3250 H 0 0 0 0 0
0.5535 3.3193 -0.3704 H 0 0 0 0 0
-1.3717 -1.1301 1.6858 H 0 0 0 0 0
-1.5810 -0.5013 -1.3080 H 0 0 0 0 0
2.6160 1.6280 0.5609 H 0 0 0 0 0
3.5948 -1.8804 -0.5097 O 0 0 0 0 0
4.1240 -1.1431 -0.8632 H 0 0 0 0 0
4.2686 -2.5619 -0.3423 H 0 0 0 0 0
-0.2472 0.9616 3.3577 O 0 0 0 0 0
0.3575 0.6108 2.6999 H 0 0 0 0 0
-1.1224 1.0508 2.9734 H 0 0 0 0 0
1 4 1 0 0
1 13 1 0 0
2 3 1 0 0
2 5 1 0 0
3 7 1 0 0
3 10 1 0 0
3 14 1 0 0
4 5 1 0 0
4 11 1 0 0
4 15 1 0 0
5 9 1 0 0
5 16 1 0 0
6 11 1 0 0
6 17 1 0 0
7 18 1 0 0
8 10 1 0 0
8 19 1 0 0
9 12 1 0 0
9 20 1 0 0
9 21 1 0 0
10 11 1 0 0
10 22 1 0 0
11 23 1 0 0
12 24 1 0 0
25 26 1 0 0
25 27 1 0 0
28 29 1 0 0
28 30 1 0 0
M END
[Execution from Interface]
Open the aDglucose_2H2O.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears.
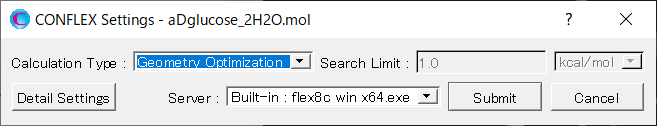
Next, in [General Settings] dialog of the detailed settings dialog, select [Host Ligand Search] from the pull-down menu of [Calculation Type:].
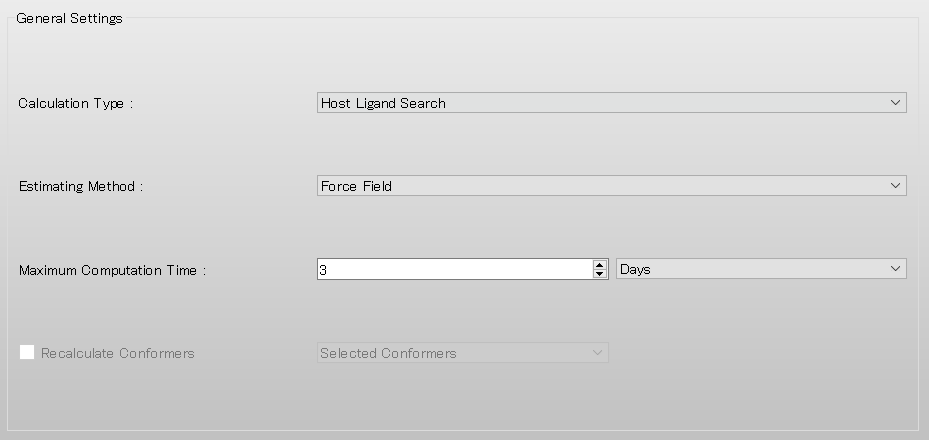
Next, configure the Host-Ligand coordination search settings in the [Host Ligand Search] dialog.
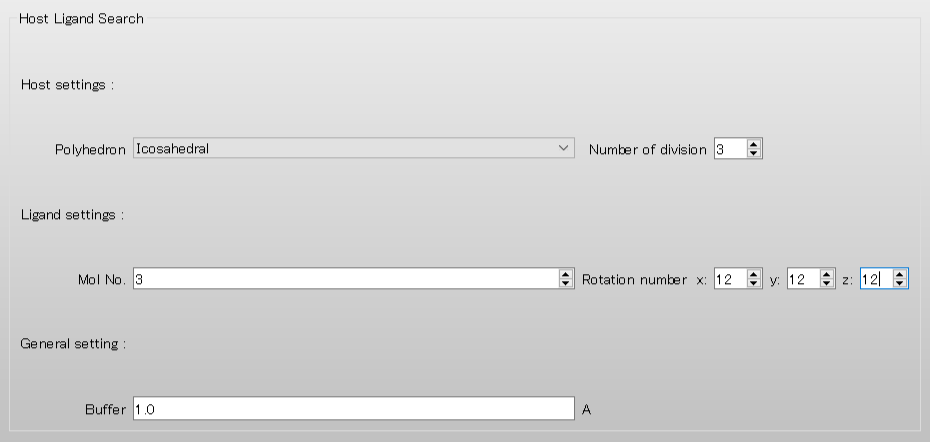
Set [Number of Division] to 3. This setting allows for a more detailed coordination search by increasing the number of ligand placement points.
Set [Mol No.] to 3 to treat the additional second water molecule as the ligand.
Additionally, set Rotation Number [x:], [y:], and [z:] each to 12 to rotate the ligand in 30-degree steps.
After completing the calculation settings, click .
Add [OPT=GROUP], [MOL_GROUP=(25,1)] and [MOL_GROUP=(28,1)] to the dialog that appears.
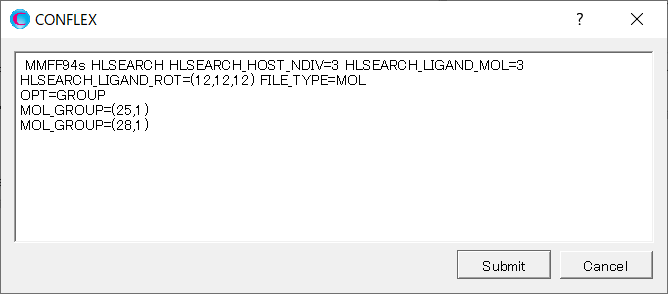
The settings [OPT=GROUP], [MOL_GROUP=(25,1)], and [MOL_GROUP=(28,1)] indicate that the positions and geometries of the water molecules are optimized, while those of α-D-glucose are kept fixed during the calculation.
For more information on geometry optimization with constraints, please refer to [Constraint by molecular object method].
After completing the modifications, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the aDglucose_2H2O.ini file.
aDglucose_2H2O.ini file
HLSEARCH OPT=GROUP MOL_GROUP=(25,1) MOL_GROUP=(28,1) HLSEARCH_HOST_NDIV=3 HLSEARCH_LIGAND_ROT=(12,12,12) HLSEARCH_LIGAND_MOL=3
Explanations of each keyword are shown below.
| Keyword | Explanation |
|---|---|
| HLSEARCH | Execute a Host-Ligand coordination search |
|
OPT=GROUP MOL_GROUP=(25,1) MOL_GROUP=(28,1) |
The positions and geometries of the water molecules are optimized, while those of α-D-glucose are kept fixed during the calculation. |
| HLSEARCH_HOST_NDIV=3 | This setting allows for a more detailed coordination search by increasing the number of ligand placement points. |
| HLSEARCH_LIGAND_ROT=(12,12,12) | The ligand is rotated in 30-degree steps. |
| HLSEARCH_LIGAND_MOL=3 | The additional second water is defined as the ligand. |
Store the two files of aDglucose_2H2O.mol and aDglucose_2H2O.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par aDglucose_2H2Oenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
The most stable structure of α-D-glucose and two water molecules found by the search is shown below.