結晶構造探索
ここでは、CONFLEXのインストール場所の
CONFLEX/Sample_Files/CONFLEX/crystal/search
フォルダ内の「tartronicacid.mol」ファイルを例として説明します。
このファイルをご自身のホームディレクトリー以下の適当な場所にコピーして作業を行なってください。
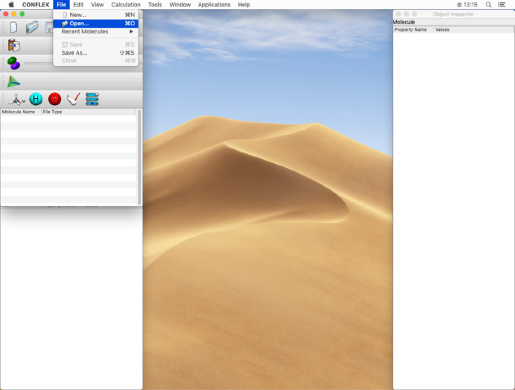
- 「File」メニューから「Open」をクリックし、コピーした「tartronicacid.mol」ファイルを開きます。
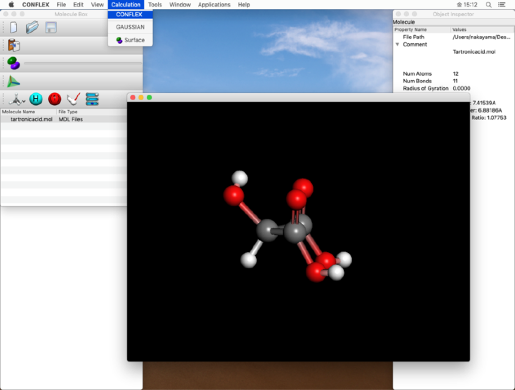
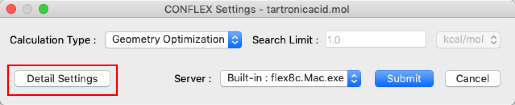
- 「Calculation」メニューから「CONFLEX」を選択し、 CONFLEX Settingsダイアログを表示させます。
- CONFLEX Settingsダイアログの「Detail Settings」をクリックし、詳細設定ダイアログを表示させます。
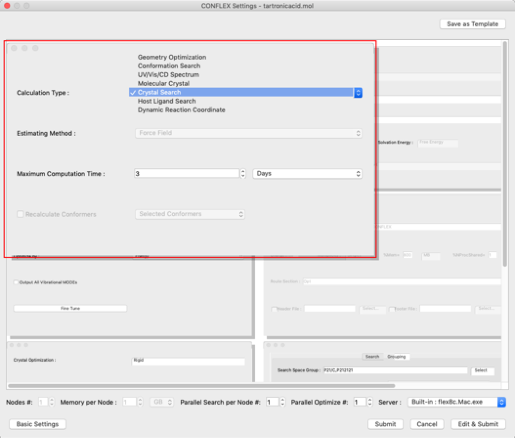
- まず、計算の種類を設定します。
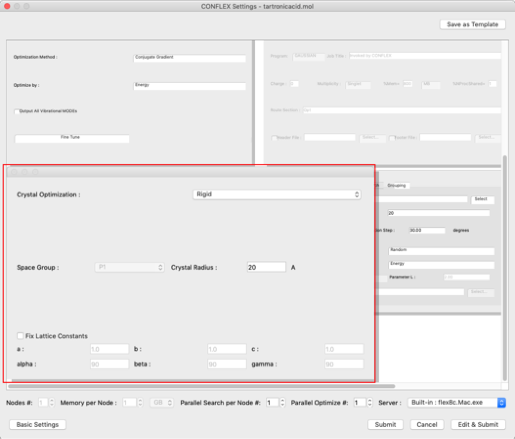
- 次に、最適化方法を設定します。
- 次に、探索方法を右下の「Crystal Search」ダイアログで設定します。
- Rotation Method:
- Grid
- Rotation Step:
- 30.00
- Position Prediction Method:
- Random
- Trial Structures:
- 20
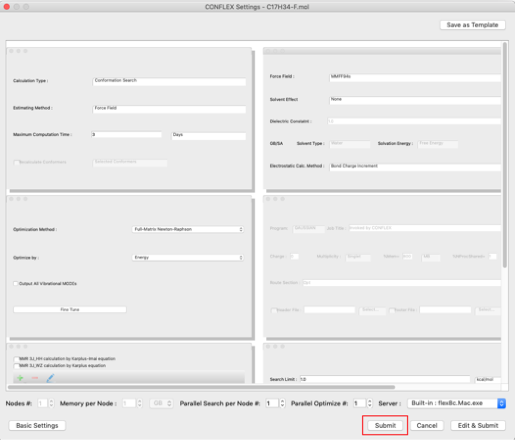
- 詳細設定ダイアログの「Submit」をクリックします。
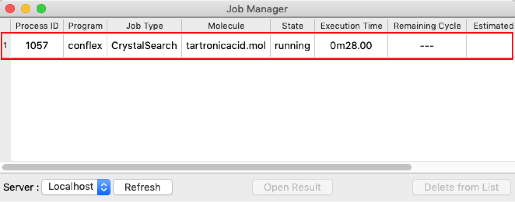
- ジョブが実行されると「Job Manager」が表示されます。
-
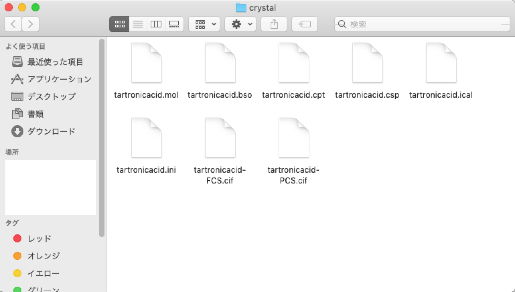
フォルダーには、「tartronicacid.mol」の他に7つのファイル
- tartronicacid.bso
- tartronicacid.cpt
- tartronicacid.csp
- tartronicacid.ical
- tartronicacid.ini
- tartronicacid-FCS.cif
- tartronicacid-PCS.cif
が含まれています。
左上にある「General Settings」ダイアログの「Calculation Type:」プルダウンメニューから「Crystal Search」を選択します。
左下の「Crystal Optimization」ダイアログの「Crystal Optimization:」プルダウンメニューから「Rigid」を選択します。
以下の各項目を設定します:
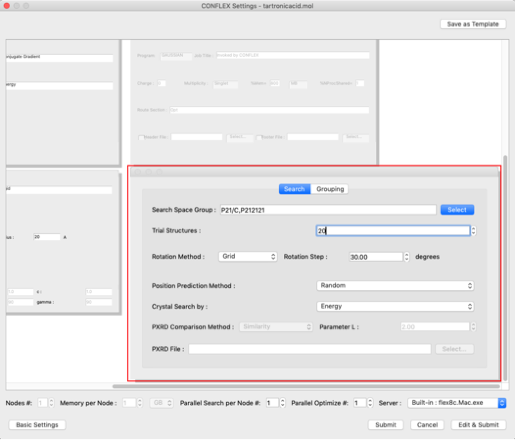
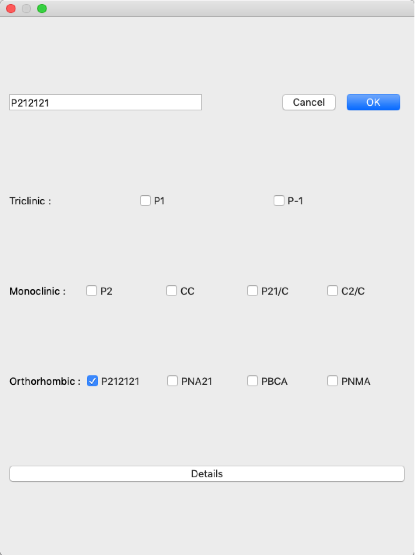
探索の空間群を「P212121」に設定します。
「Search Space Group:」の「Select」をクリックし、表示されるダイアログから「P212121」をチェックします。
それ以外のチェックボックスがチェックされていないことを確認して、「OK」をクリックします。
結晶構造探索ジョブが実行されます。
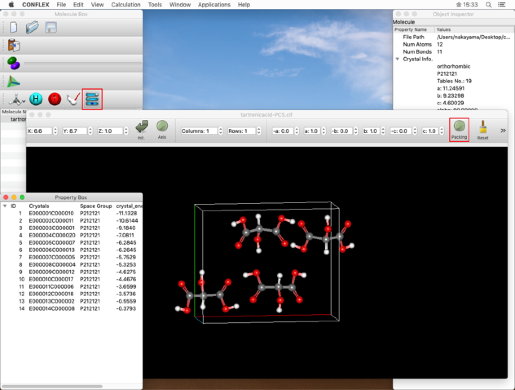
「State」が「Finished」(ジョブ終了)となっていることを確認したら、赤枠部分をダブルクリックします。
ダブルクリックによって開く分子ウィンドウには、探索により得られた結晶構造が表示されます。その他の結晶構造は、「Property Box」ダイアログにリストされており、エネルギー値をクリックすることで、その結晶構造が分子ウィンドウに表示されます。
パッキング構造を表示する場合、Molecule BoxダイアログのControllerアイコン をクリックし、分子ウィンドウに表示されたツールバーにある「Packing」ボタンをクリックします。
をクリックし、分子ウィンドウに表示されたツールバーにある「Packing」ボタンをクリックします。