その場配向探索
分子が複数存在する系において、対象分子が他の分子に対してどのような向きでいるのが安定であるかを求められれば、溶液中の溶質ー溶媒間や、生体高分子と低分子との相互作用を求める上で有用です。
CONFLEXに搭載された「その場配向探索(In-Situ Orientation Search)」機能により、対象分子を入力構造の位置で順次回転させて初期構造を作り、それぞれ構造最適化することで配向の異なる複合体構造を得ることができます。

対象分子を回転させる角度は「ISOSEARCH_LIGAND_ROT=(l,m,n)」で指定でき、デフォルトでは360/6=60°刻みです(l=m=n=6)。これを例えば45°刻みにする場合は「ISOSEARCH_LIGAND_ROT=(8,8,8)」とします。
【α-シクロデキストリン+フェノール複合体の探索】
下図のように、α-シクロデキストリンの内部近くにフェノール分子を置いた構造データ(aCD_phenol.mol)が、CONFLEXがインストールされているフォルダー内のSample_Files/CONFLEX/in-situ_searchフォルダー内に用意されていますので、これをコピーしてCONFLEX Interfaceで開きます。尚、下図のように水素原子を省略して表示するには、CONFLEX InterfaceのViewメニューからShow/Hide -> Hydrogensを選択します。
ここでは、フェノール分子について最適な配向を探索する計算を行います。
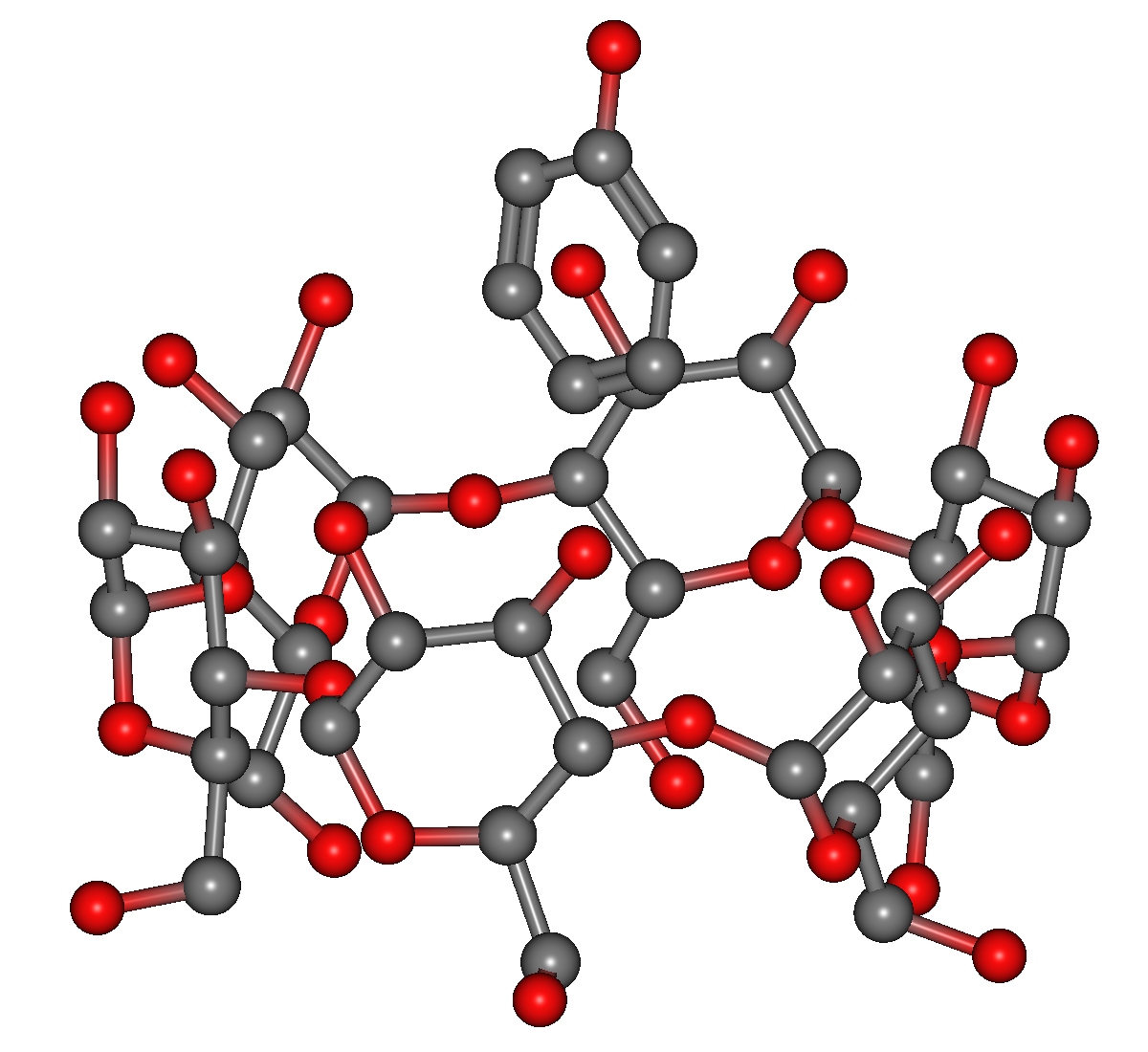
α-シクロデキストリン+フェノールの入力構造データ(aCD_phenol.mol)
aCD_phenol.mol
139145 0 0 0 1 V2000
-0.9900 -5.1813 1.7326 C 0 0 0 0 0
0.4703 -5.2557 2.1169 C 0 0 0 0 0
1.3126 -4.4420 1.1593 C 0 0 0 0 0
1.0559 -4.8370 -0.2876 C 0 0 0 0 0
-0.4459 -4.8668 -0.5733 C 0 0 0 0 0
...
...
3.3185 3.0495 3.8277 H 0 0 0 0 0
1 2 1 0 0 0
1 10 1 6 0 0
1 20 1 0 0 0
...
...
131137 1 6 0 0
132138 1 0 0 0
133139 1 0 0 0
M END
[Interfaceから実行する場合]
Calculationメニューから「CONFLEX」を選択して、開いた計算設定ダイアログのをクリックします。詳細設定ダイアログが表示されますので、「General Settings」ダイアログにある「Calculation Type:」 のプルダウンメニューから「Host Ligand Search」を選択します。
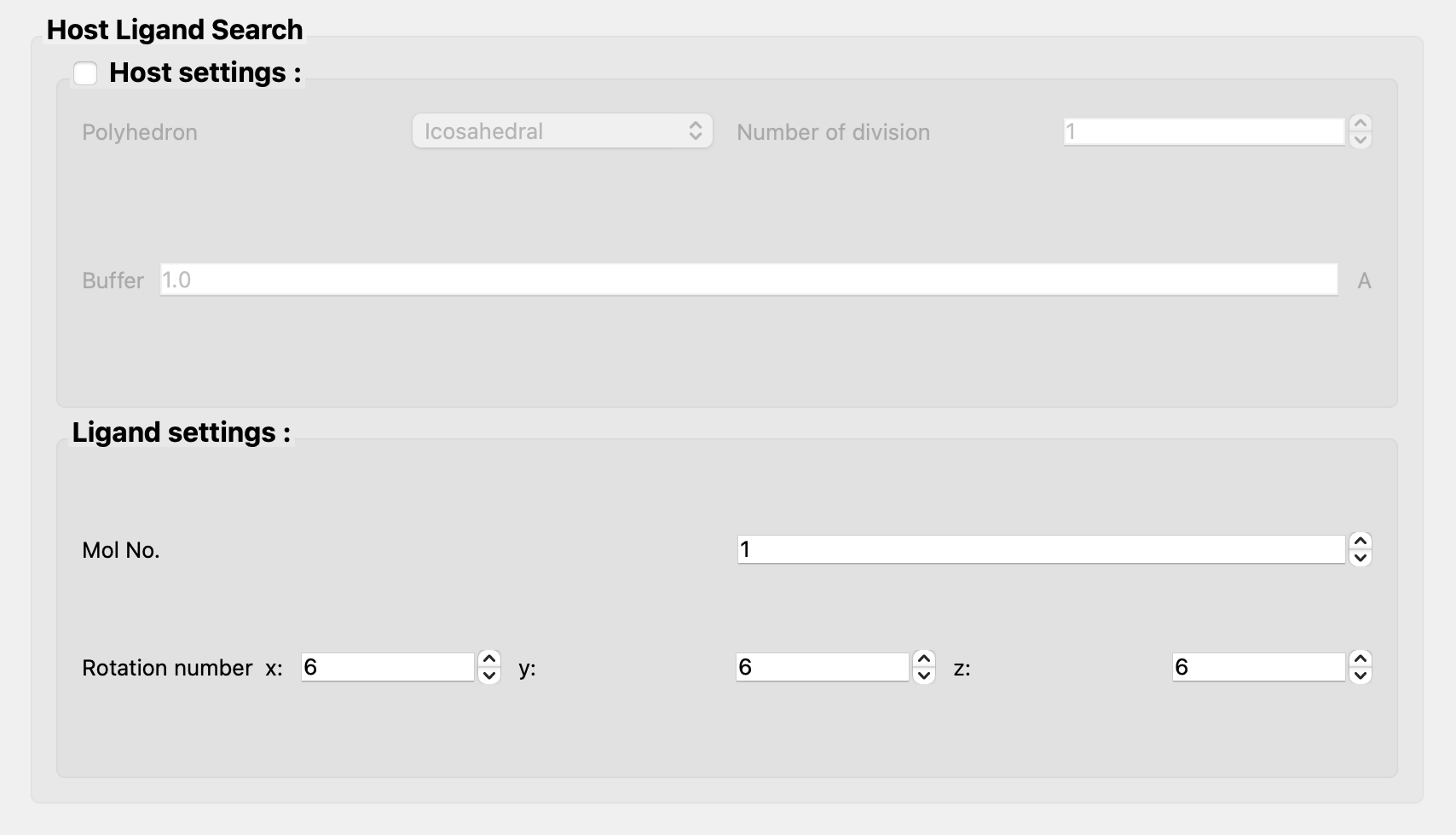
次に、「Host Ligand Search」ダイアログにある「Host settings :」のチェックを外します。

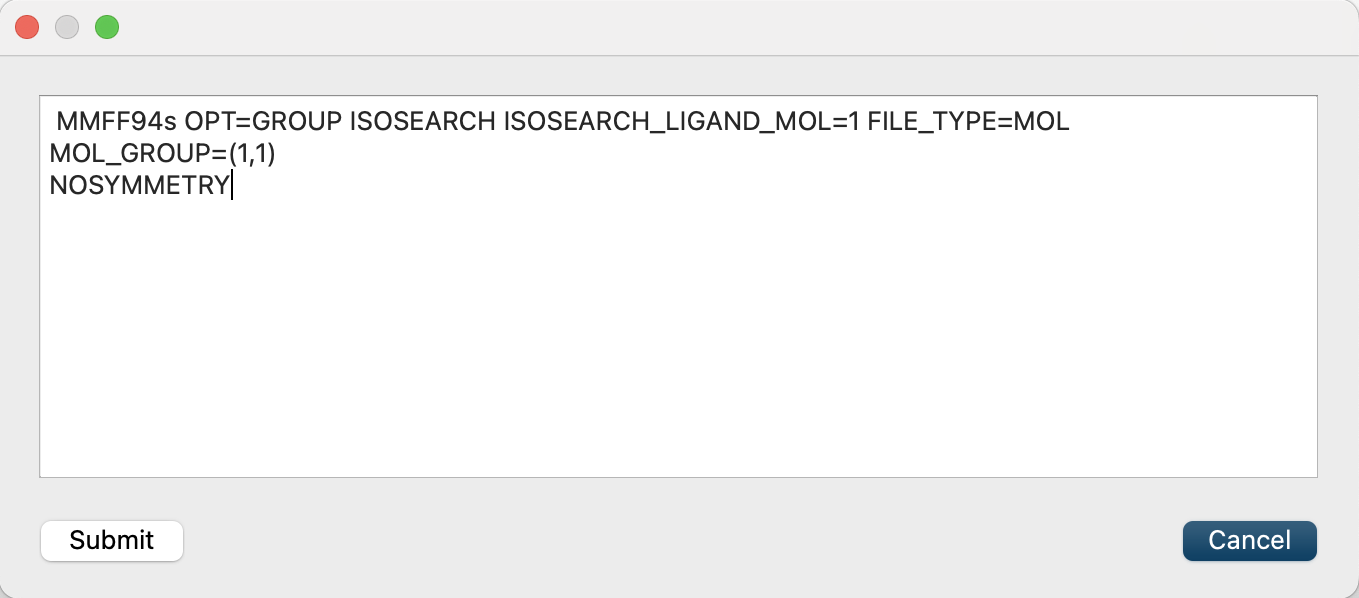
最後に、をクリックして、開いたダイアログに「NOSYMMETRY」を追記します。これにより、対称性を判定するための構造変換を行わないようになります。
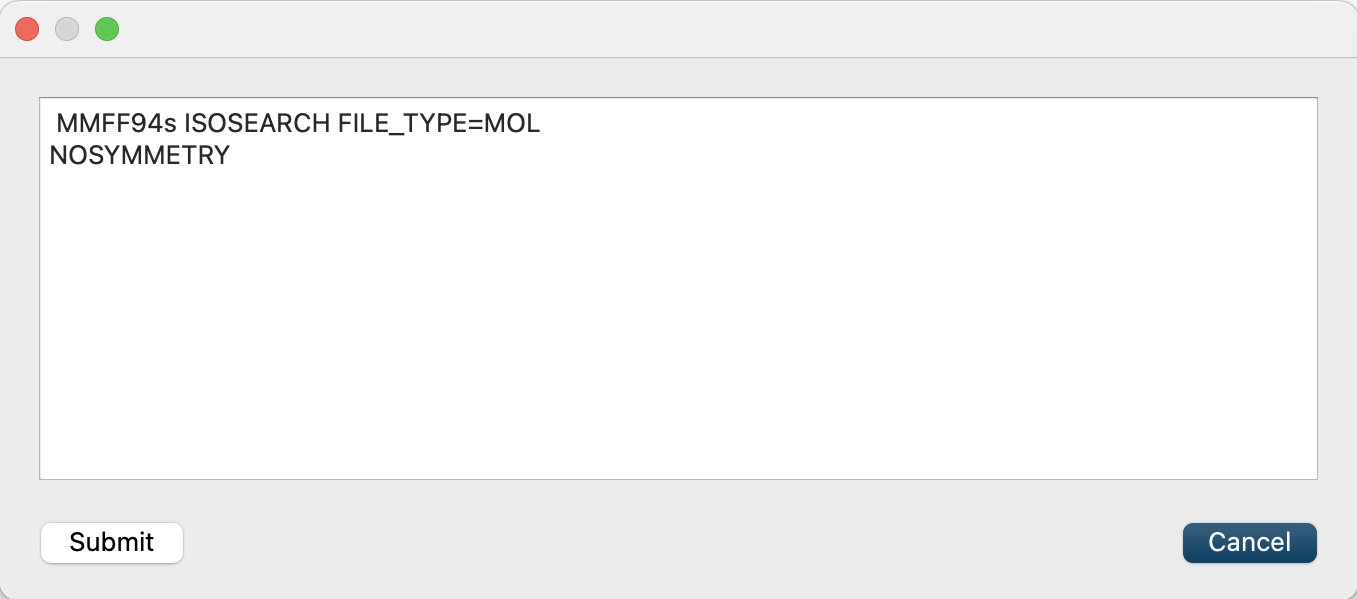
設定が終わりましたら、をクリックします。フェノール分子の配向探索が行われます。
[コマンドラインから実行する場合]
aCD_phenol.iniファイルを用意し、以下のキーワードを記述して保存します。
aCD_phenol.iniファイル
ISOSEARCH NOSYMMETRY
「ISOSEARCH」キーワードを含めることで、その場配向探索が行われます。
aCD_phenol.molとaCD_phenol.iniを同じフォルダーに格納し、下記のコマンドを実行してください。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par aCD_phenolenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
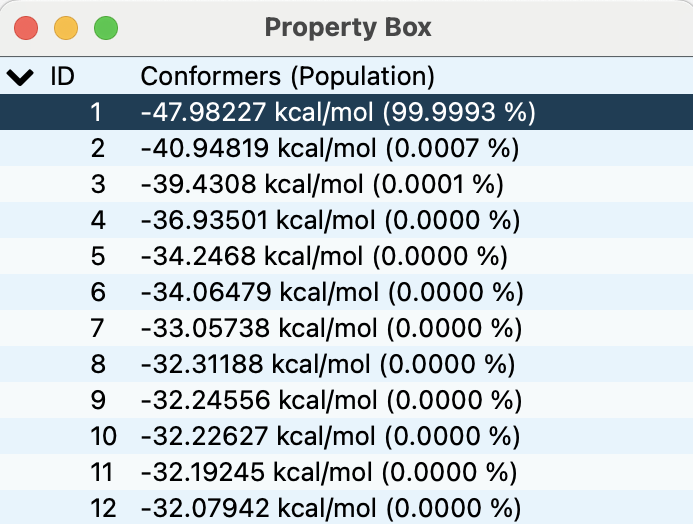
計算結果
上記の計算により、56個の構造が得られました。最安定構造を下図に示します。
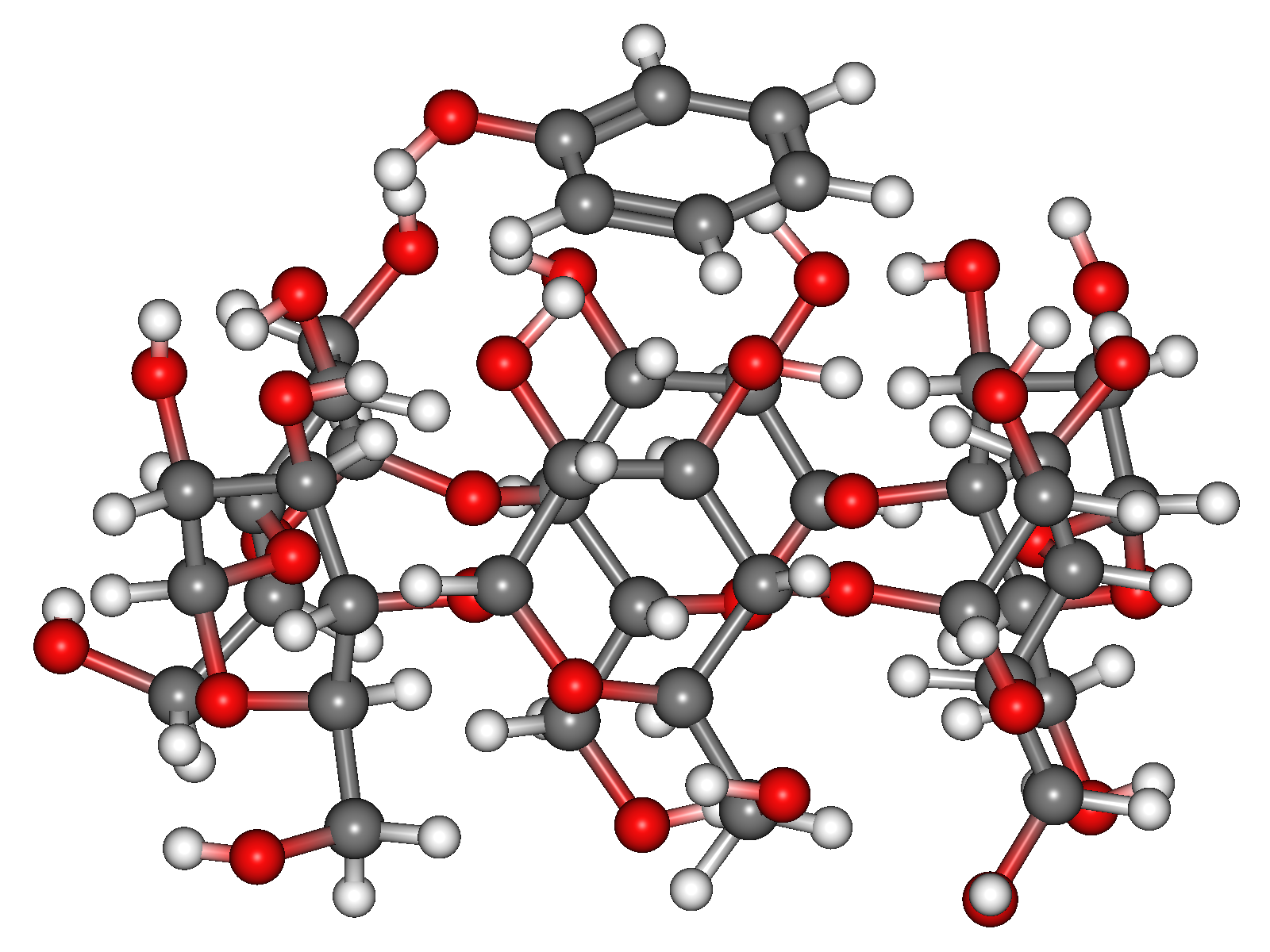
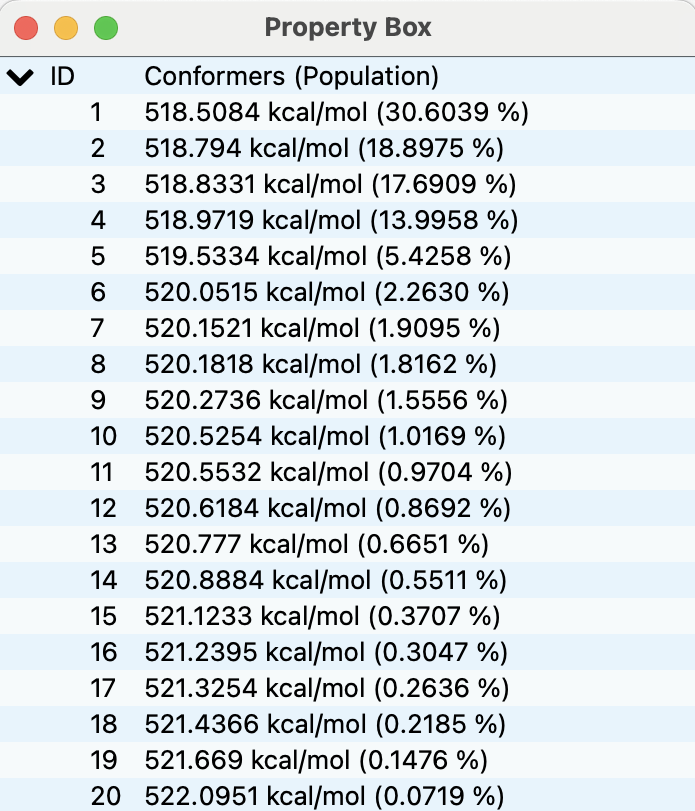
またエネルギー値のリストは、配座探索計算と同様Property Boxに表示されます(下図)。
可視化の方法は、「計算結果の可視化」をご覧ください。
【酢酸3分子系の探索】
酢酸3分子を、下図のように中心分子とその左右(Z軸方向)に5Å平行に離した構造データ(acetic_acid_trimer.mol)が、CONFLEXがインストールされているフォルダー内のSample_Files/CONFLEX/in-situ_searchフォルダー内に用意されていますので、これをコピーしてCONFLEX Interfaceで開きます。
ここでは、両端の分子を固定し、中央の分子について最適な配向を探索する計算を行います。
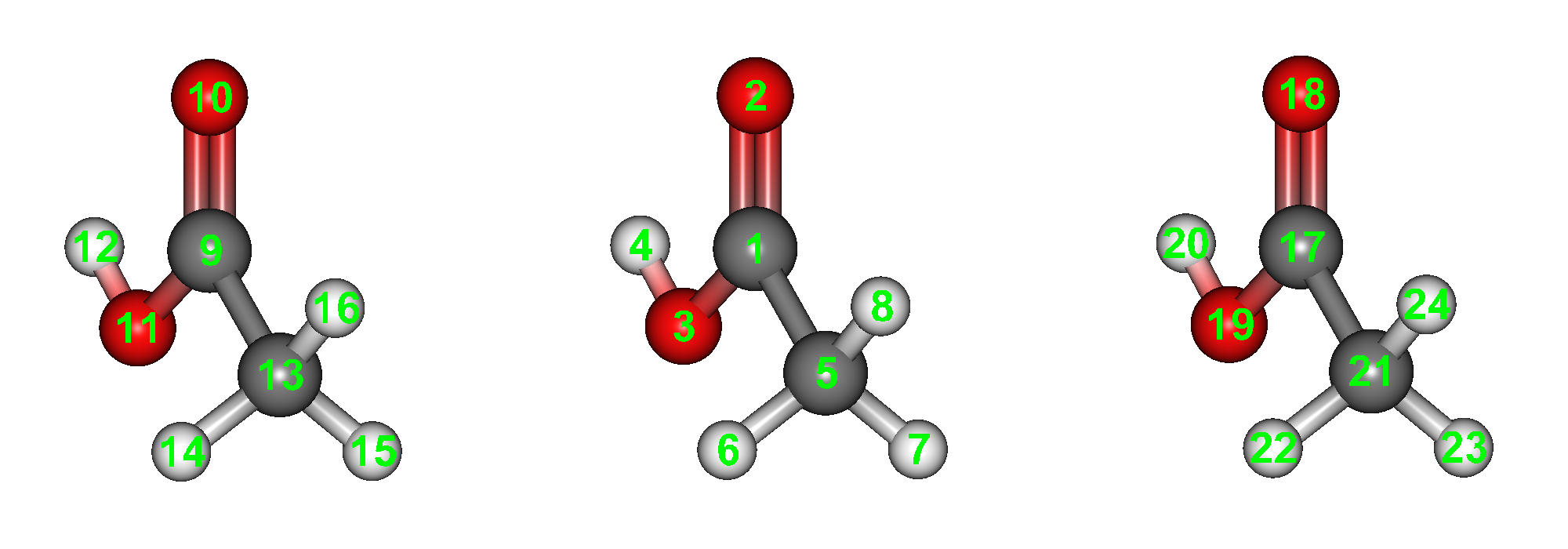
酢酸3分子のの入力構造データ(acetic_acid_trimer.mol)
acetic_acid_trimer.mol
24 21 0 0 0 0 0 0 0 0 0 0
0.9628 -0.5511 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1931 -0.5511 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.3335 0.6511 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
...
...
0.5138 -2.6519 -5.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0 0 0 0
1 3 1 0 0 0 0
1 5 1 0 0 0 0
...
...
21 24 1 0 0 0 0
M END
[Interfaceから実行する場合]
Calculationメニューから「CONFLEX」を選択して、開いた計算設定ダイアログのをクリックします。詳細設定ダイアログが表示されますので、「General Settings」ダイアログにある「Calculation Type:」 のプルダウンメニューから「Host Ligand Search」を選択します。
次に、「Geometry Optimization」ダイアログにある「Optimization Method :」プルダウンメニューから「Group Optimization」を選択します。
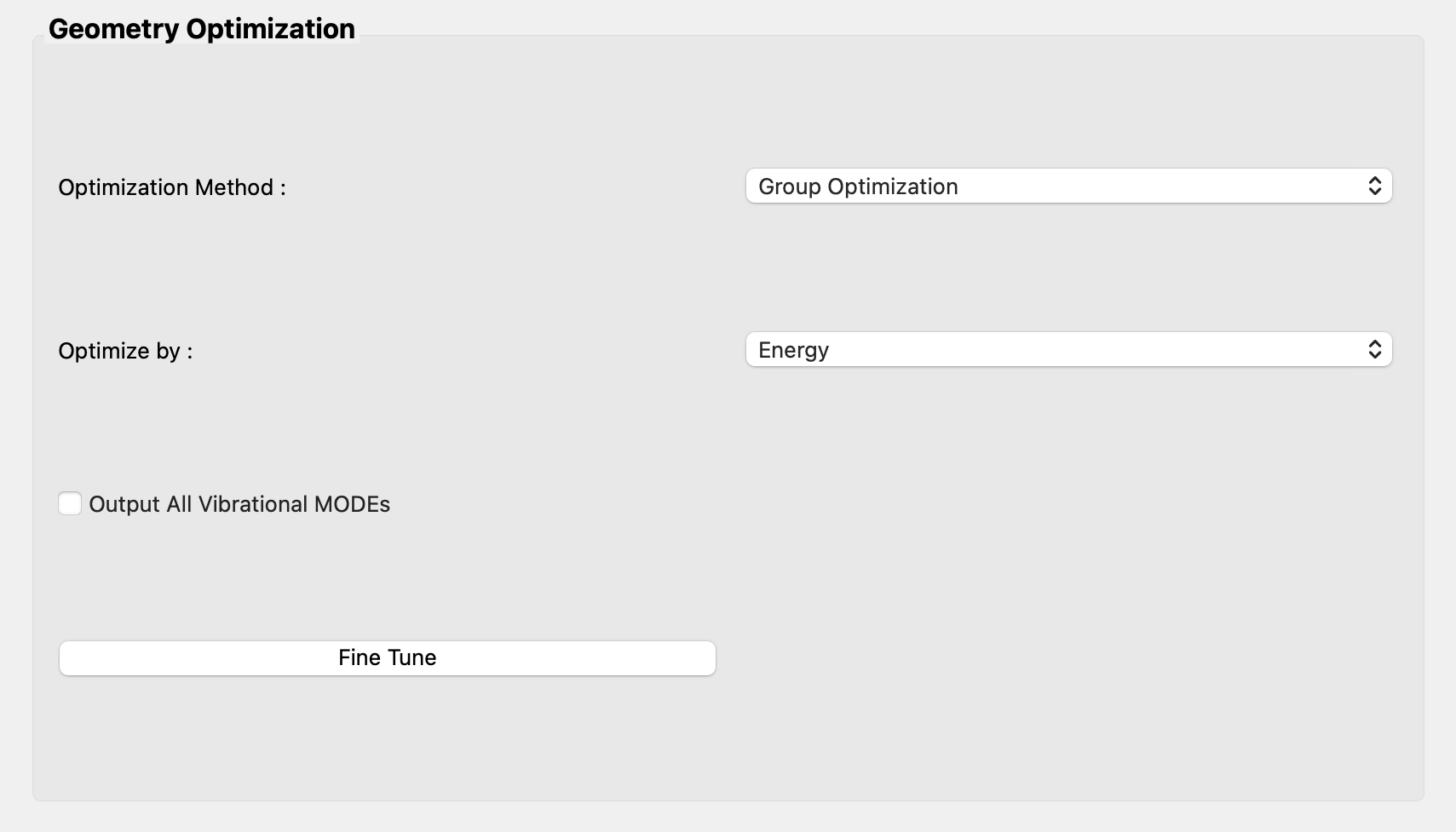
次に、「Host Ligand Search」ダイアログにある「Host settings :」のチェックを外し、「Ligand settings :」の「Mol No.」を「1」にします。
最後に、をクリックして、開いたダイアログに「MOL_GROUP=(1,1)」および「NOSYMMETRY」を追記します。これにより、中央の分子のみが最適化され、また対称性を判定するための構造変換を行わないようになります。
[コマンドラインから実行する場合]
acetic_acid_trimer.iniファイルを用意し、以下のキーワードを記述して保存します。
acetic_acid_trimer.iniファイル
ISOSEARCH ISOSEARCH_LIGAND_MOL=1 OPT=GROUP MOL_GROUP=(1,1) NOSYMMETRY
「ISOSEARCH」キーワードを含めることでその場配向探索が行われ、「ISOSEARCH_LIGAND_MOL=1」で中央の分子を探索対象としています。また、「OPT=GROUP」および「MOL_GROUP=(1,1)」とすることで、中央の分子のみ構造最適化を行うようにします。
部分固定に関しては、「構造を部分的に固定した計算 → 分子単位での構造固定」をご覧ください。
acetic_acid_trimer.molとacetic_acid_trimer.iniを同じフォルダーに格納し、下記のコマンドを実行してください。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par acetic_acid_trimerenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
上記の計算により、12個の構造が得られました。分子表示画面上部の「Columns:」を「4」、「Rows:」を「3」にすることで、以下のように3行4列に分割され、全ての構造を表示することができます。

またエネルギー値のリストは、配座探索計算と同様Property Boxに表示されます(下図)。
可視化の方法は、「計算結果の可視化」をご覧ください。