AMBER force field calculation
This section describes how to perform a geometry optimization and vibrational analysis using AMBER force field. We use TRP Cage as an example.
[Geometry optimization and vibrational analysis]
First, prepare molecular structure data of TRP Cage in Amber prmtop format and Amber inpcrd format as trpcage.prmtop and trpcage.crd.
For creating those files, use LEaP program in AmberTools. About how to use it in detail, please refer to Amber manual and tutorial.
The amino acid sequence of TRP Cage is [NLYIQWLKDGGPSSGRPPPS] in one letter code. You should create the LEap input file (file name: tleap.in) containing the following.
source leaprc.protein.ff14SB
TRP = sequence { NASN LEU TYR ILE GLN TRP LEU LYS ASP GLY GLY PRO SER SER GLY ARG PRO PRO PRO CSER }
saveAmberParm TRP trpcage.prmtop trpcage.crd
quit
After that, execute tLEap by the following command. The trpcage.prmtop and trpcage.crd will be generated. Note that you should set up the environment setting for AMBER program in advance.
tleap -f tleap.inenter
You can find the trpcage.prmtop and trpcage.crd in Sample_Files folder in the folder installed CONFLEX (Sample_Files\CONFLEX\amber\trpcage.[prmtop|crd]).
About file formats of prmtop and crd files, please refer to Amber file formats. Regarding the prmtop file in detail, please also refer to prmtop.pdf.
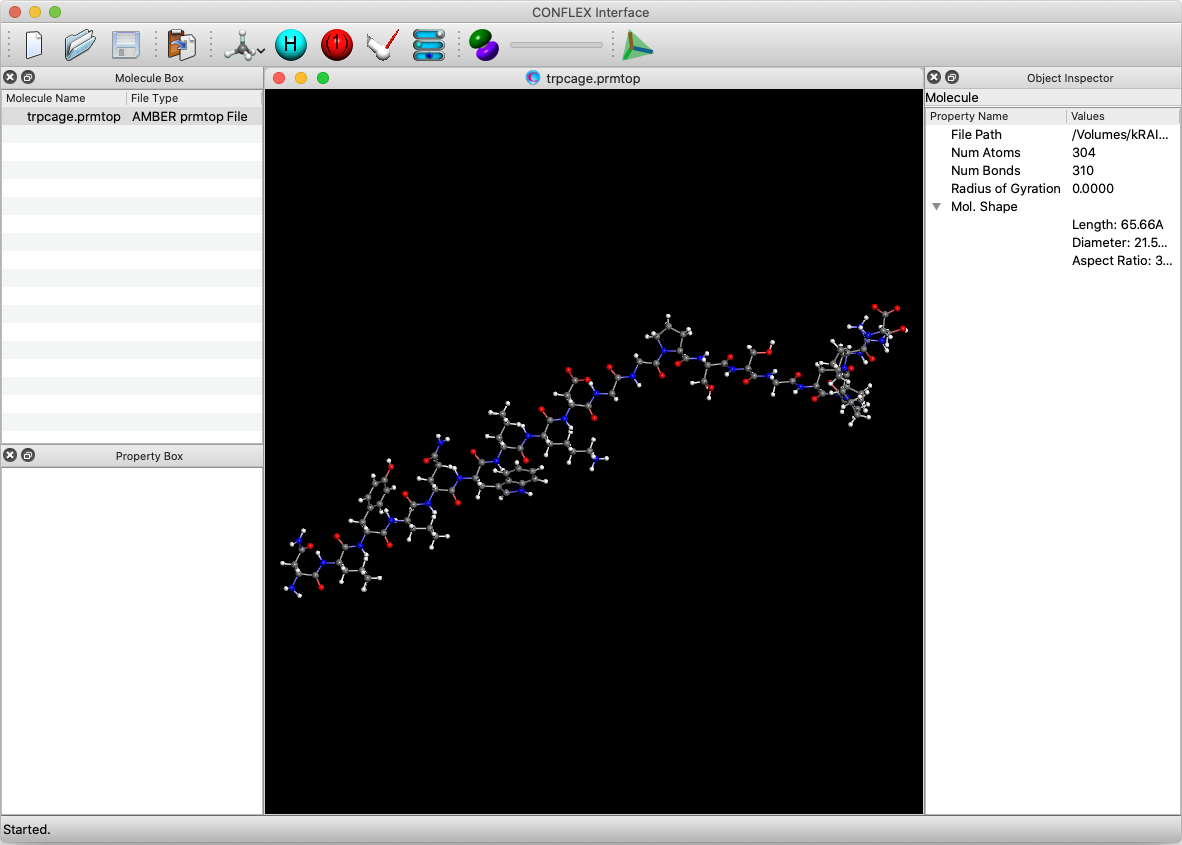
The first part of contents in the trpcage.prmtop file is shown below. It contains the number of atoms, the number of residues, the atomic name assigned to each atom, and so on. The contents in trpcage.prmtop file are not changed in the simulation.
%VERSION VERSION_STAMP = V0001.000 DATE = 12/01/20 16:04:37
%FLAG TITLE
%FORMAT(20a4)
NASN
%FLAG POINTERS
%FORMAT(10I8)
304 12 150 160 346 219 700 656 0 0
1701 20 160 219 656 53 124 138 26 0
0 0 0 0 0 0 0 0 24 0
0
%FLAG ATOM_NAME
%FORMAT(20a4)
N H1 H2 H3 CA HA CB HB2 HB3 CG OD1 ND2 HD21HD22C O N H CA HA
CB HB2 HB3 CG HG CD1 HD11HD12HD13CD2 HD21HD22HD23C O N H CA HA CB
HB2 HB3 CG CD1 HD1 CE1 HE1 CZ OH HH CE2 HE2 CD2 HD2 C O N H CA HA
CB HB CG2 HG21HG22HG23CG1 HG12HG13CD1 HD11HD12HD13C O N H CA HA CB
HB2 HB3 CG HG2 HG3 CD OE1 NE2 HE21HE22C O N H CA HA CB HB2 HB3 CG
〜〜〜〜〜〜〜〜〜〜
The first part of contents in the trpcage.crd file is shown below. It contains the molecular name, the number of atoms, and the x, y, and z coordinates of each atom.
NASN 304 3.3257700 1.5479090 -0.0000016 4.0461540 0.8399910 -0.0000029 2.8230940 1.4995080 -0.8746870 2.8230970 1.4995070 0.8746850 3.9700480 2.8457950 -0.0000001 3.6716630 3.4001290 -0.8898200 3.5769650 3.6538380 1.2321430 2.4969950 3.8010750 1.2413790 3.8774840 3.1157950 2.1311970 4.2537000 5.0171120 1.2321440 5.0052990 5.3404060 0.3150720 3.9848850 5.8179090 2.2659170 4.4080150 6.7337020 2.3147430 3.3596110 5.5042970 2.9944640 5.4855410 2.7052070 -0.0000044 6.0088240 1.5931750 -0.0000084 〜〜〜〜〜〜〜〜〜〜
[Execution from Interface]
First, open the trpcage.prmtop file using the CONFLEX Interface. When opening the file, select [AMBER prmtop file] as the file format. Since the prmtop file does not contain coordinate data, the dialog will appear again when clicking . In the dialog that appears, select the trpcage.crd containing the coordinate data and click .
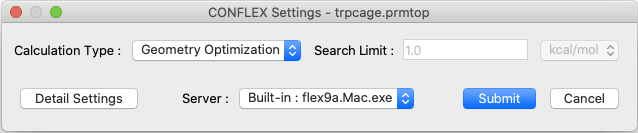
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. This will perform molecular geometry optimization and normal mode analysis. If the molecular file is in Amber format, the Interface automatically select the Amber force field.
[Execution via command line]
Store the two files of trpcage.prmtop and trpcage.crd in a single folder, and then execute command below to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par trpcageenter
The command above is for Windows OS. For other operating systems, please refer to [How to execute CONFLEX].
[Output files for geometry optimization]
After the calculation is completed, the following files are primarily output.
- trpcage.bso
- This file contains the force field parameters used in the calculation, energies of each interaction as well as thermodynamic quantities, frequencies, and vibrational modes obtained from the normal mode analysis, etc.
- trpcage-F.mol2
- This files contains the optimized structure in Sybyl-Mol2 format.
- trpcage-F.prmtop
- The contents of this file match those of the trpcage.prmtop file.
- trpcage-F.crd
- This files contains the optimized structure in AMBER Inpcrd format.
[If you executed the calculation using the command line]
You can find the trpcage.bso file in the folder where the input files are stored. You can visualize the optimized structure by opening the bso file using the CONFLEX Interface.