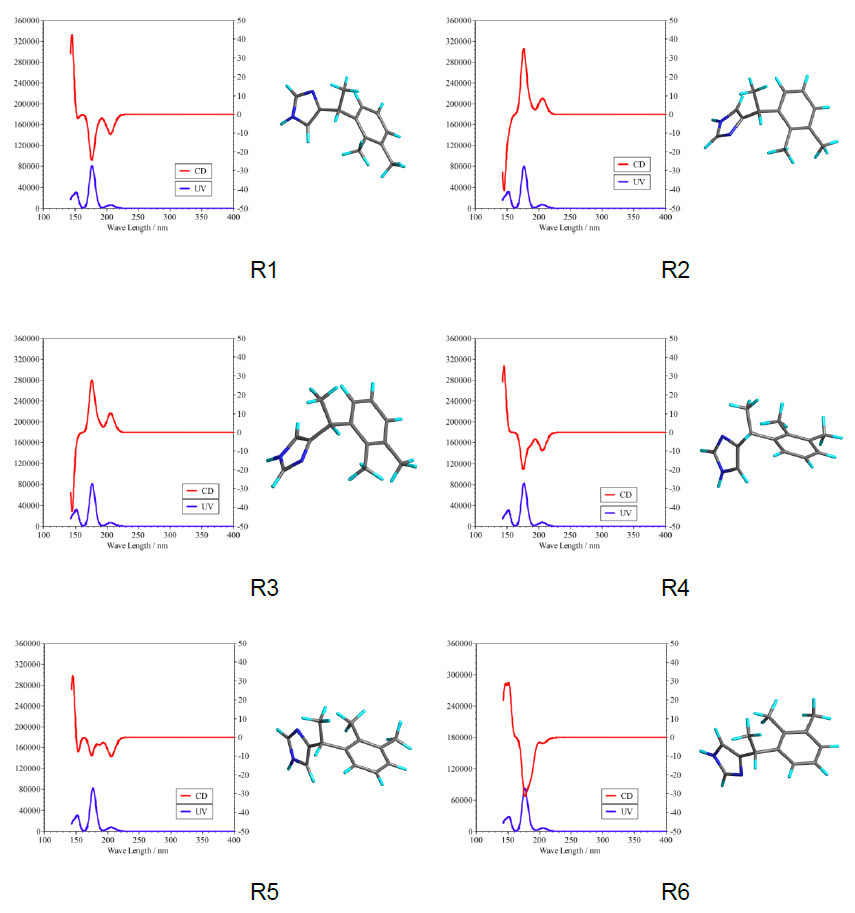
CD/UV spectrum analysis
Dexmedetomidine is a medetomidine activator (D-form), featuring an imidazole skeleton, and acts as a highly potent and selective central α2 adrenaline receptor agonist.
By stimulating central α2 receptors, it inhibits sympathetic nerve signal transmission and produces a sedative effect.
Additionally, this molecule exhibits the simplest form of optical activity.
This section explains how to perform CD/UV spectrum calculations using the dexmedetomidine molecule.
Structural formula of dexmedetomidine: Precedex®
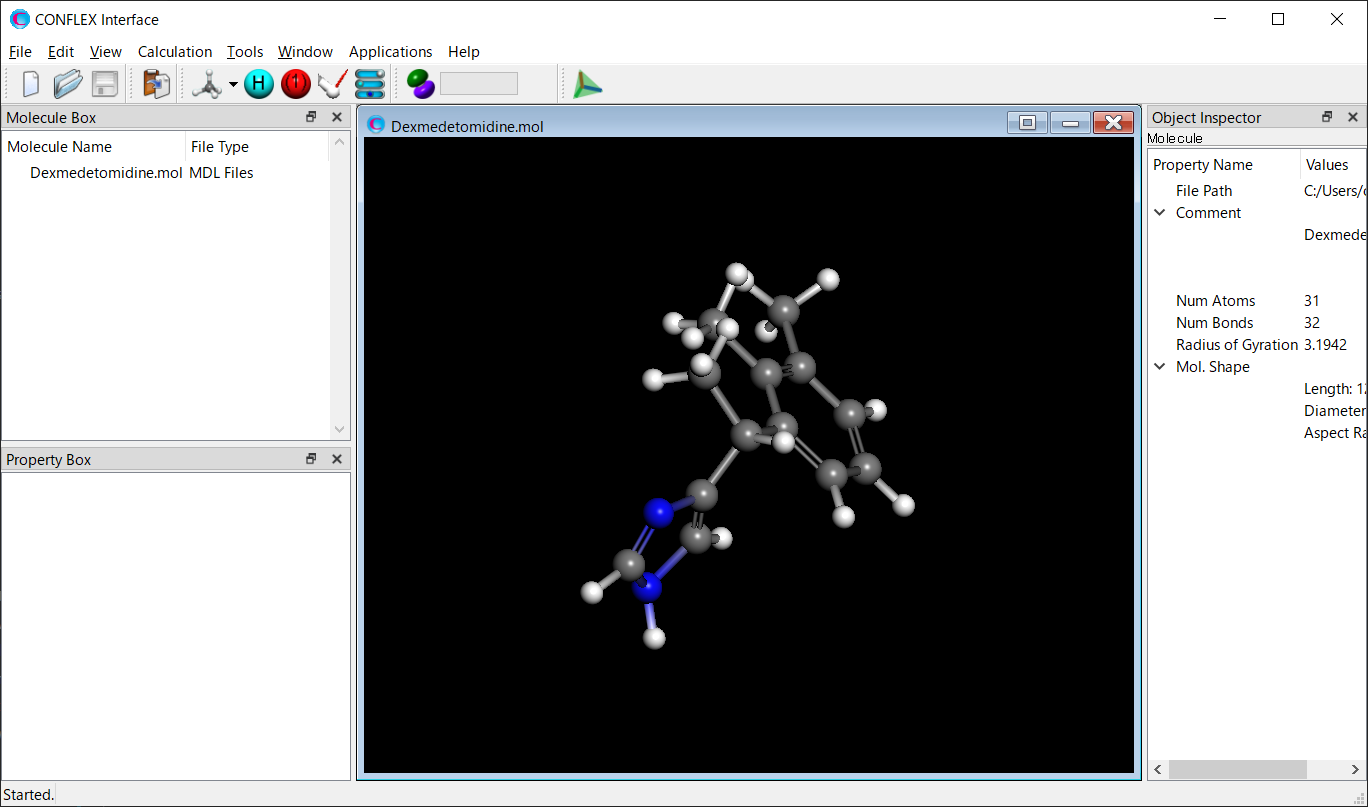
Structure data of dexmedetomidine (R-form) (Dexmedetomidine.mol)
Dexmedetomidine
31 32 0 0 0 1 V2000
-1.0778 -1.6715 0.6897 C 0 0 0 0 0
-2.0603 -2.6647 1.0836 N 0 0 0 0 0
-2.4168 -2.2186 2.4226 C 0 0 0 0 0
-1.8248 -1.1779 2.8306 N 0 0 0 0 0
-0.9540 -0.8156 1.7107 C 0 0 0 0 0
-0.0612 0.3840 1.7794 C 0 0 0 0 0
0.6530 0.5305 0.4721 C 0 0 0 0 0
1.6443 -0.4148 0.0972 C 0 0 0 0 0
2.3211 -0.2864 -1.1434 C 0 0 0 0 0
2.0041 0.7900 -2.0119 C 0 0 0 0 0
1.0134 1.7374 -1.6414 C 0 0 0 0 0
0.3382 1.6082 -0.3982 C 0 0 0 0 0
-0.8947 1.6284 2.0582 C 0 0 0 0 0
-0.7060 2.6067 -0.0050 C 0 0 0 0 0
0.6790 2.8690 -2.5617 C 0 0 0 0 0
-0.5680 -1.6802 -0.2851 H 0 0 0 0 0
-1.9104 -3.6810 0.8775 H 0 0 0 0 0
-3.1582 -2.7716 3.0179 H 0 0 0 0 0
0.6828 0.2465 2.5956 H 0 0 0 0 0
1.8873 -1.2500 0.7705 H 0 0 0 0 0
3.0881 -1.0203 -1.4318 H 0 0 0 0 0
2.5261 0.8903 -2.9749 H 0 0 0 0 0
-0.9541 1.8063 3.1554 H 0 0 0 0 0
-1.9247 1.4948 1.6581 H 0 0 0 0 0
-0.4306 2.5163 1.5733 H 0 0 0 0 0
-1.1426 2.3320 0.9812 H 0 0 0 0 0
-0.2528 3.6205 0.0707 H 0 0 0 0 0
-1.5176 2.6296 -0.7662 H 0 0 0 0 0
-0.1300 3.4917 -2.1188 H 0 0 0 0 0
1.5781 3.5043 -2.7251 H 0 0 0 0 0
0.3338 2.4705 -3.5419 H 0 0 0 0 0
1 2 1 0 0 0
1 5 2 0 0 0
1 16 1 6 0 0
2 3 1 1 0 0
2 17 1 0 0 0
3 4 2 0 0 0
3 18 1 1 0 0
4 5 1 6 0 0
5 6 1 0 0 0
6 7 1 6 0 0
6 13 1 0 0 0
6 19 1 1 0 0
7 8 2 0 0 0
7 12 1 6 0 0
8 9 1 6 0 0
8 20 1 1 0 0
9 10 2 0 0 0
9 21 1 0 0 0
10 11 1 0 0 0
10 22 1 6 0 0
11 12 2 0 0 0
11 15 1 6 0 0
12 14 1 0 0 0
13 23 1 1 0 0
13 24 1 0 0 0
13 25 1 0 0 0
14 26 1 1 0 0
14 27 1 0 0 0
14 28 1 6 0 0
15 29 1 0 0 0
15 30 1 0 0 0
15 31 1 6 0 0
M END
Conformation search
[Execution from Interface]
Open the Dexmedetomidin.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
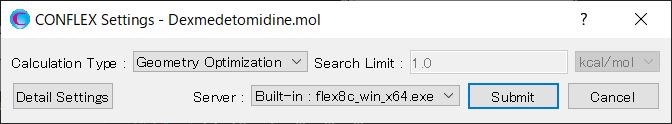
Select [Conformation Search] from the [Calculation Type:] pull-down menu in [General Settings] dialog of the detailed settings dialog.
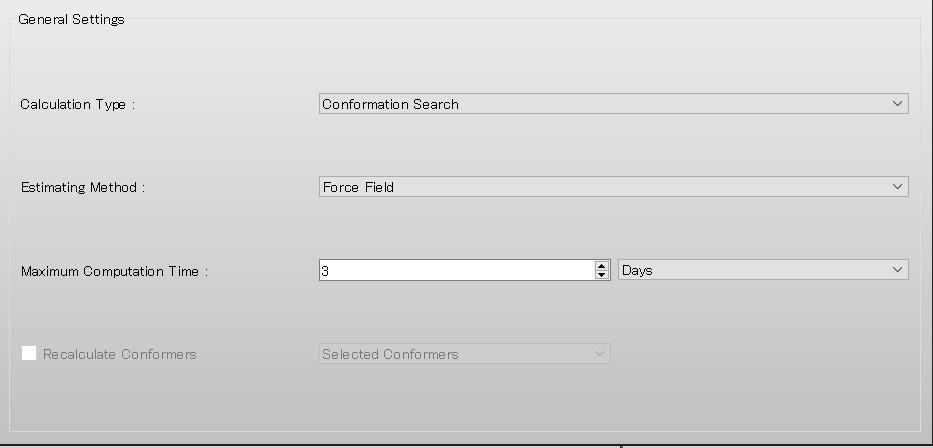
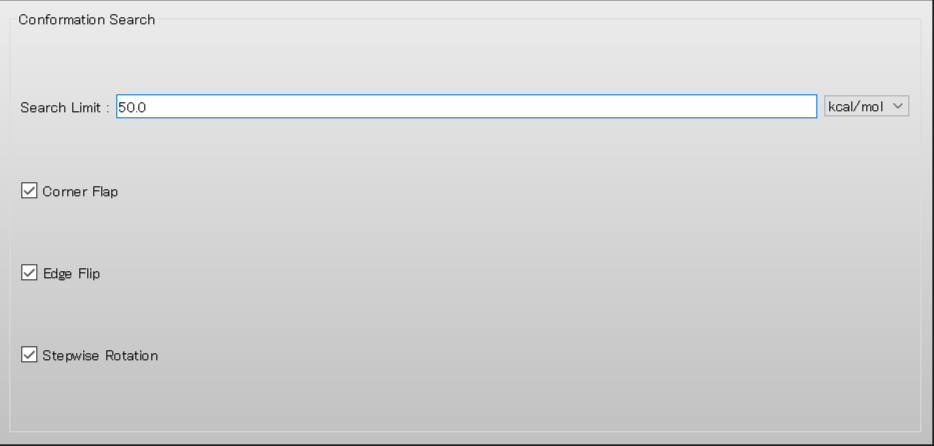
Edit the value of [Search Limit:] to 50.0 in [Conformation Search] dialog of the detailed settings dialog.
When completing the calculation settings, click .
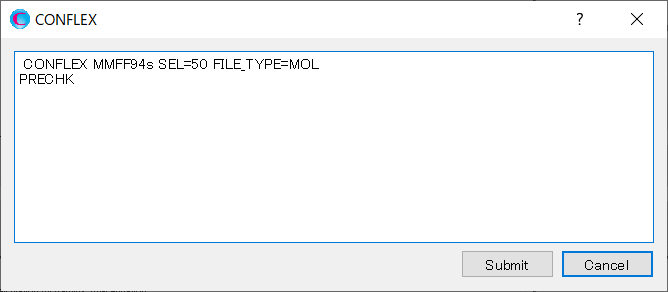
Add [PRECHK] to the dialog that appears, and then click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the Dexmedetomidin.ini file.
Dexmedetomidin.ini file
MMFF94S CONFLEX PRECHK SEL=50.0
- [MMFF94S] means to use MMFF94s force field.
- [CONFLEX] means to perform a conformation search.
- [SEL=50.0] means to set a search limit to 50.0 kcal/mol.
Store the two files of Dexmedetomidin.mol and Dexmedetomidin.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par Dexmedetomidinenter
The above command is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
We can obtaine six conformers from the conformation search.
| No.ID | Conf ID. | Steric E (kcal/mol) | ΔE (kcal/mol) | Distribution (%) |
|---|---|---|---|---|
| R1 | 2 | 48.8457 | 0 | 47.0162 |
| R2 | 5 | 49.1525 | 0.3068 | 28.0111 |
| R3 | 3 | 49.2567 | 0.411 | 23.4962 |
| R4 | 1 | 51.2498 | 2.4041 | 0.8129 |
| R5 | 4 | 51.3723 | 2.5266 | 0.6611 |
| R6 | 6 | 54.6699 | 5.8242 | 0.0025 |
CD/UV spectrum calculation
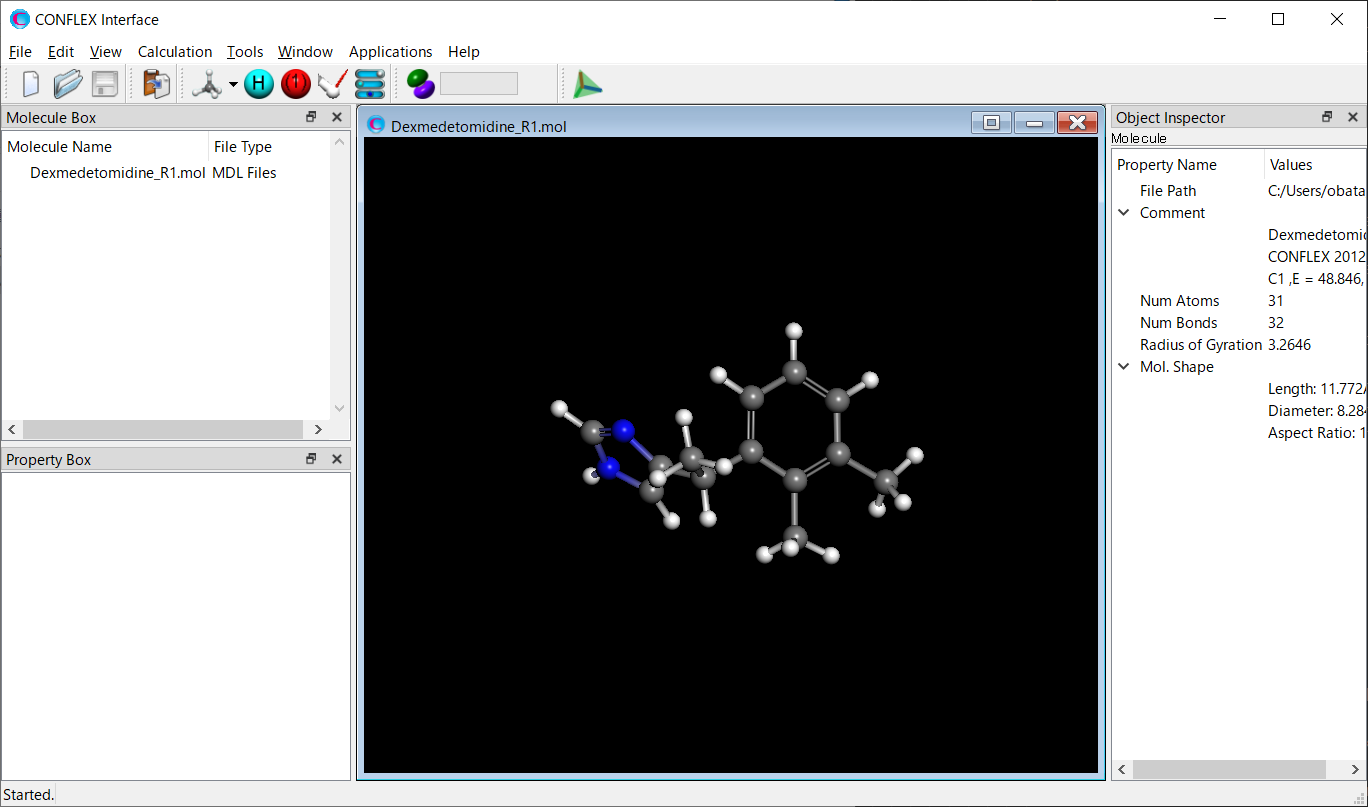
CD/UV spectrum calculations are performed for the conformers. The structure data for conformers R1–R6 are stored in the Dexmedetomidine.sdf in MDL MOL file format. Here, we extract the structure data of the R1 conformer from Dexmedetomidine.sdf and save it as Dexmedetomidine_R1.mol.
Structure data of R1 conformer (Dexmedetomidine_R1.mol)
Dexmedetomidine
CONFLEX 20120911103D 1 1.00000 48.84570 2
C1 ,E = 48.846, G = 0.585E-07, P = 47.0162, M( 0), IFN =00000001-00000002
31 32 0 0 1 V2000
-2.0874 -0.9516 -1.0953 C 0 0 0 0 0
-3.2087 -0.3537 -1.5970 N 0 0 0 0 0
-3.6187 0.5800 -0.6906 C 0 0 0 0 0
-2.8252 0.6169 0.3538 N 0 0 0 0 0
-1.8462 -0.3288 0.1149 C 0 0 0 0 0
-0.7355 -0.5857 1.0969 C 0 0 0 0 0
0.5101 0.0794 0.5067 C 0 0 0 0 0
0.5314 1.4826 0.3608 C 0 0 0 0 0
1.6413 2.1406 -0.1565 C 0 0 0 0 0
2.7595 1.4107 -0.5382 C 0 0 0 0 0
2.7854 0.0130 -0.4098 C 0 0 0 0 0
1.6575 -0.6664 0.1168 C 0 0 0 0 0
-1.0377 -0.1065 2.5179 C 0 0 0 0 0
1.6676 -2.1687 0.2694 C 0 0 0 0 0
4.0321 -0.7132 -0.8424 C 0 0 0 0 0
-1.5600 -1.7306 -1.6269 H 0 0 0 0 0
-3.6565 -0.5595 -2.4790 H 0 0 0 0 0
-4.4935 1.1972 -0.8447 H 0 0 0 0 0
-0.6109 -1.6715 1.1573 H 0 0 0 0 0
-0.3396 2.0732 0.6439 H 0 0 0 0 0
1.6304 3.2216 -0.2630 H 0 0 0 0 0
3.6185 1.9431 -0.9407 H 0 0 0 0 0
-1.9187 -0.6210 2.9181 H 0 0 0 0 0
-1.2403 0.9691 2.5673 H 0 0 0 0 0
-0.1934 -0.3152 3.1844 H 0 0 0 0 0
1.5390 -2.4397 1.3225 H 0 0 0 0 0
0.8650 -2.6129 -0.3286 H 0 0 0 0 0
2.5987 -2.6334 -0.0623 H 0 0 0 0 0
3.8018 -1.4182 -1.6475 H 0 0 0 0 0
4.7912 -0.0217 -1.2241 H 0 0 0 0 0
4.4752 -1.2463 0.0048 H 0 0 0 0 0
1 2 1 0 0
1 5 2 0 0
1 16 1 6 0
2 3 1 1 0
2 17 1 0 0
3 4 2 0 0
3 18 1 1 0
4 5 1 6 0
5 6 1 0 0
6 7 1 6 0
6 13 1 0 0
6 19 1 1 0
7 8 2 0 0
7 12 1 6 0
8 9 1 6 0
8 20 1 1 0
9 10 2 0 0
9 21 1 0 0
10 11 1 0 0
10 22 1 6 0
11 12 2 0 0
11 15 1 6 0
12 14 1 0 0
13 23 1 1 0
13 24 1 0 0
13 25 1 0 0
14 26 1 1 0
14 27 1 0 0
14 28 1 6 0
15 29 1 0 0
15 30 1 0 0
15 31 1 6 0
M END
[Execution from Interface]
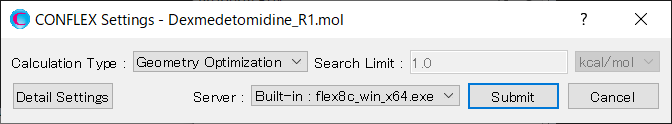
Open Dexmedetomidin_R1.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
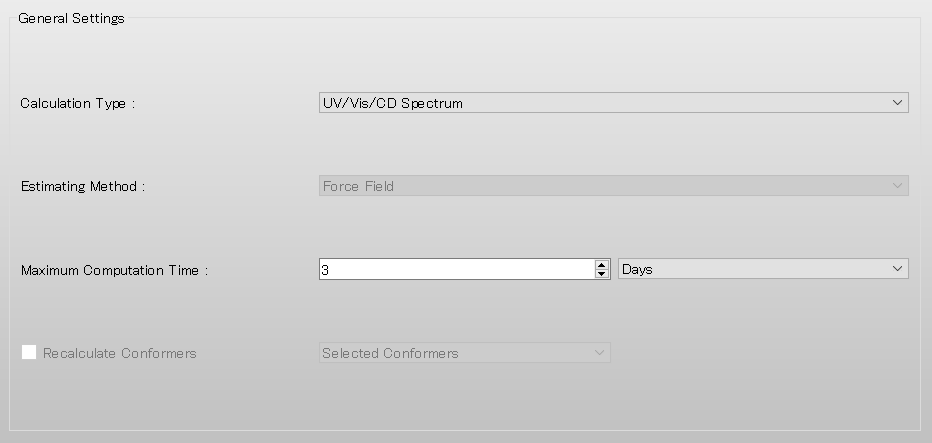
Select [UV/Vis/CD Spectrum] from the pull-down menu of [Calculation Type:] in [General Settings] dialog of the detailed settings dialog.
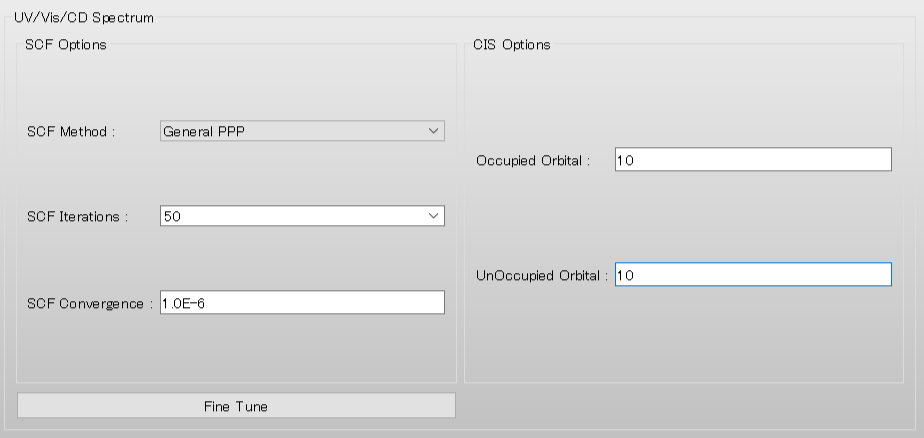
Next, we configure the parameters for the CD/UV spectrum calculation using [UV/Vis/CD Spectrum] dialog.
Select [Specify...] from the [SCF Iterations:] pull-down menu, and input 50. This sets the maximum number of SCF iterations to 50.
Set [Occupied Orbital:] and [UnOccupied Orbital:] to 10, respectively. These settings define the number of occupied orbitals (Nomo) and unoccupied orbitals (Numo) used for one-electron excited CI calculations.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the Dexmedetomidin_R1.ini file.
Dexmedetomidin_R1.ini file
MMFF94s CDUV SCF_ITER=50 CIS=(10,10)
- [MMFF94S] means to use MMFF94s force field.
- [CDUV] means to perform CD/UV spectrum calculation.
- [SCF_ITER=50] means to set the maximum number of SCF iterations to 50.
- [CIS=(10,10)] means to set the number of occupied orbitals (Nomo) and unoccupied orbitals (Numo) used for one-electron excited CI calculations to 10, respectively.
Store the two files of Dexmedetomidin_R1.mol and Dexmedetomidin_R1.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par Dexmedetomidin_R1enter
The above command is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
The results of CD/UV spectrum calculation are shown at the end of Dexmedetomidin_R1.bso file.
!---------------------------------------------------------------------------------------------------------------------!
! CURVE PLOTTING !
! SCALING FACTOR: 1.00000 !
!---------------------------------------------------------------------------------------------------------------------!
WAVELENGTH(NM) WAVENUMBER(1/CM) UV(STR) UV(VEL) CD(VEL)
1000.000 10000.0 0.00000 0.00000 0.00000
980.392 10200.0 0.00000 0.00000 0.00000
961.538 10400.0 0.00000 0.00000 0.00000
943.396 10600.0 0.00000 0.00000 0.00000
925.926 10800.0 0.00000 0.00000 0.00000
909.091 11000.0 0.00000 0.00000 0.00000
892.857 11200.0 0.00000 0.00000 0.00000
877.193 11400.0 0.00000 0.00000 0.00000
862.069 11600.0 0.00000 0.00000 0.00000
847.458 11800.0 0.00000 0.00000 0.00000
......
The graph generated from this data is shown below, along with the results for conformers R2–R6.