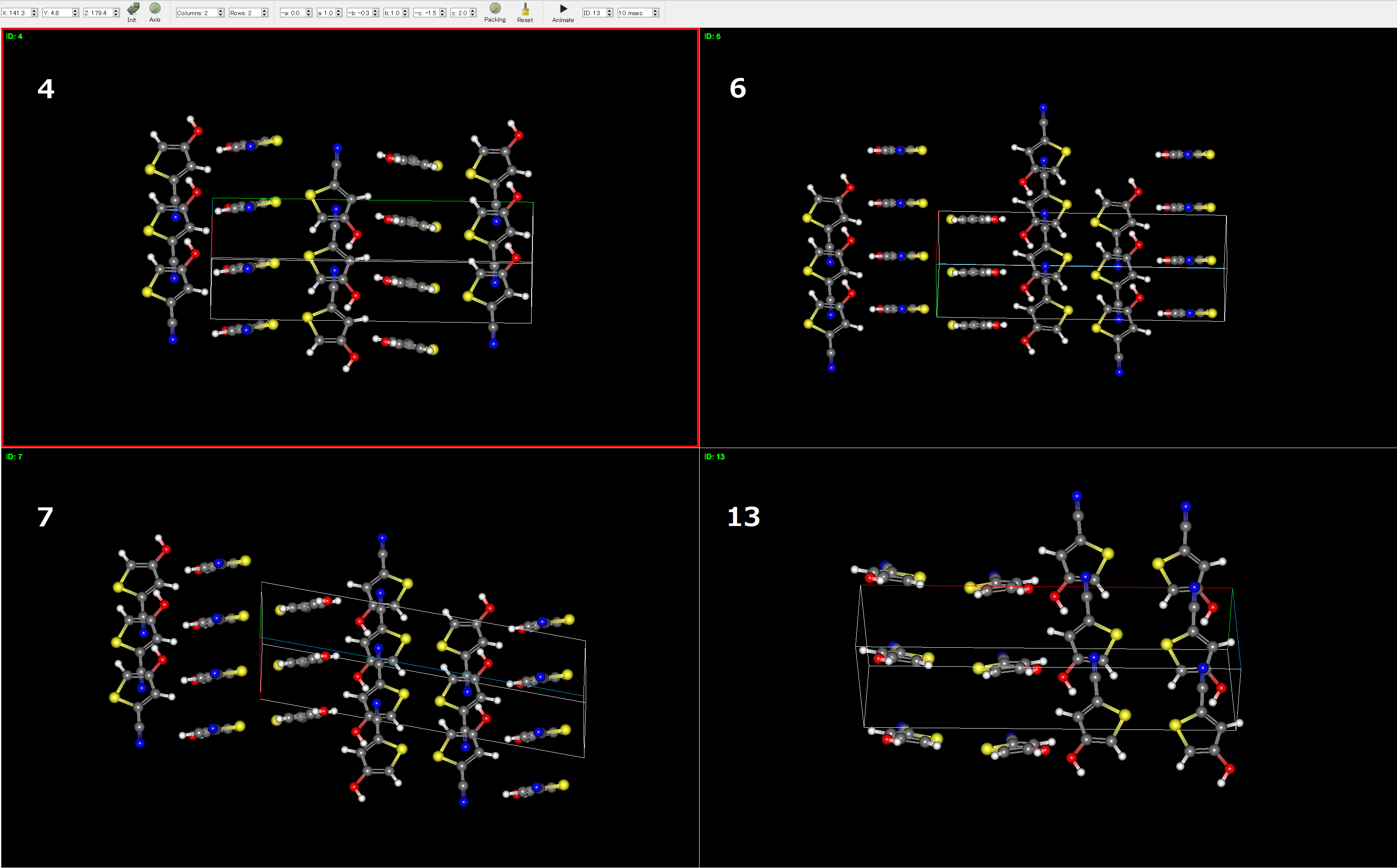
Grouping of crystal structures
CONFLEX can group structures obtained from a crystal structure search based on the similarity of their powder X-ray diffraction (PXRD) patterns.
Structures within the same group are considered to have similar atomic arrangements.
In this study, we use the results of crystal structure search for 5-Cyano-3-hydroxythiophene (II), and perform grouping of its crystal structures. The PXRD similarity calculations are carried out using Gelder's method [R. de Gelder et al, J Comput Chem 22: 273–289, 2001.].
In the case of grouping structures obtained from a previously performed crystal structure search, we utilize the restart function of the crystal structure search.
[If you executed the search using Interface]
Store the II-c2.mol and II-c2.ini files used for the search, along with the II-c2.cpt file generated after the search, in a single folder. Then, open the II-c2.mol file using the CONFLEX Interface.
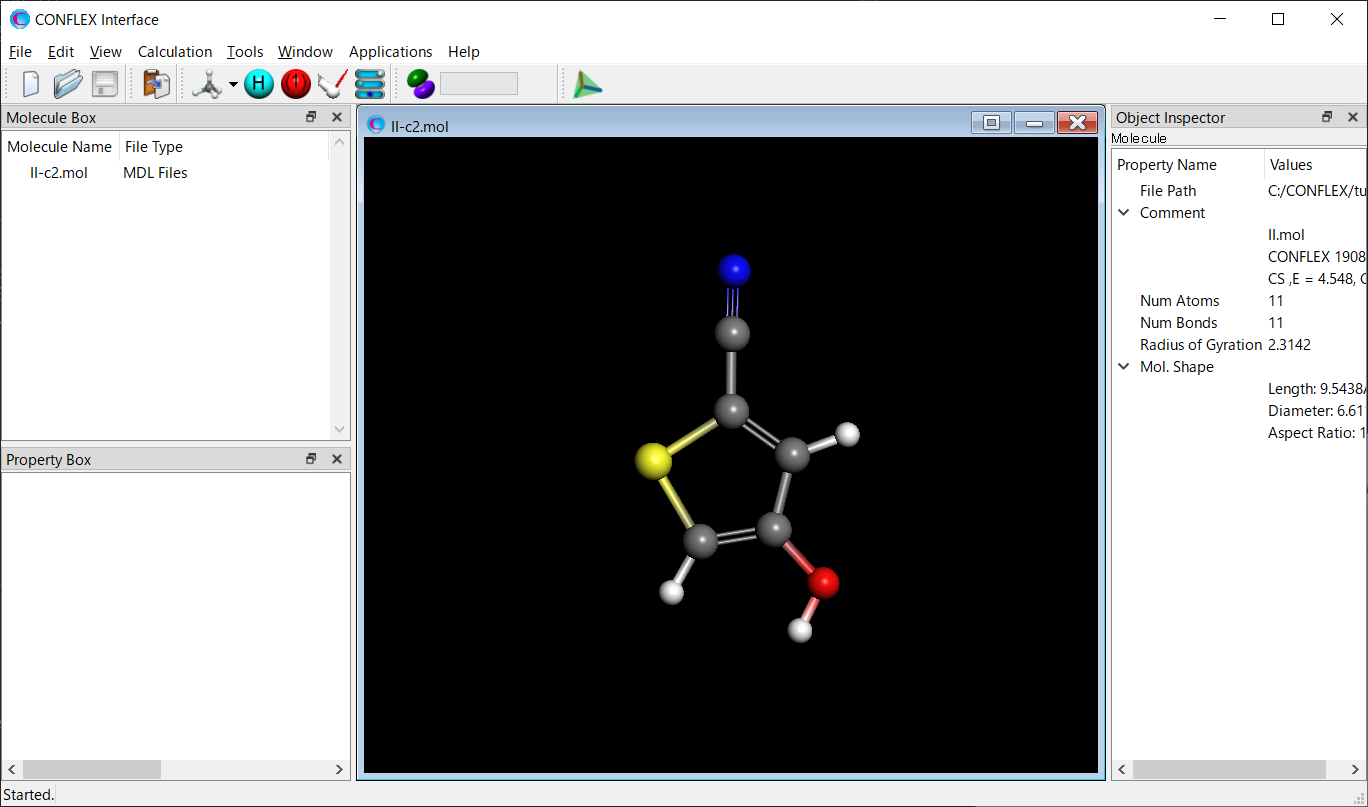
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
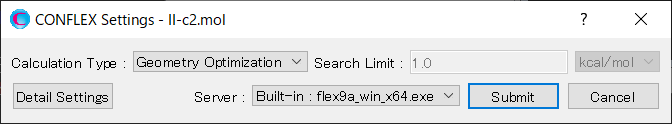
To restart the search calculation, check the [Restart calculation] checkbox located at the bottom of the [Crystal Search] dialog in the detailed settings dialog.
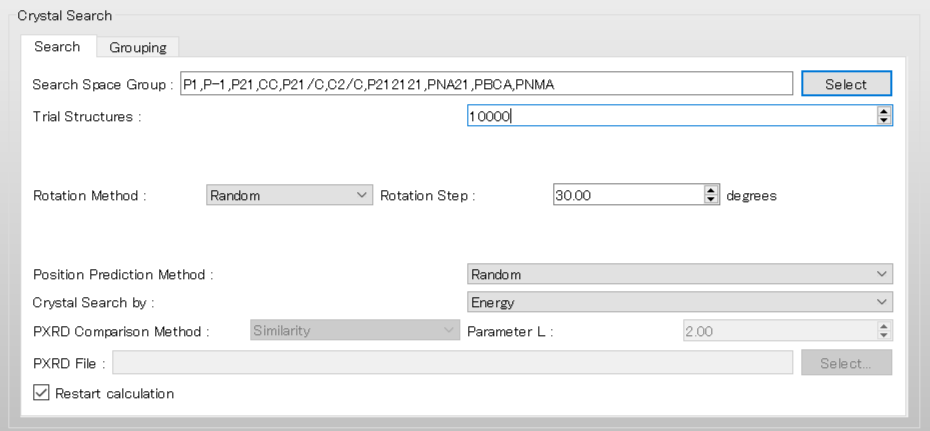
Next, select [Grouping] tab in the [Crystal Search] dialog.
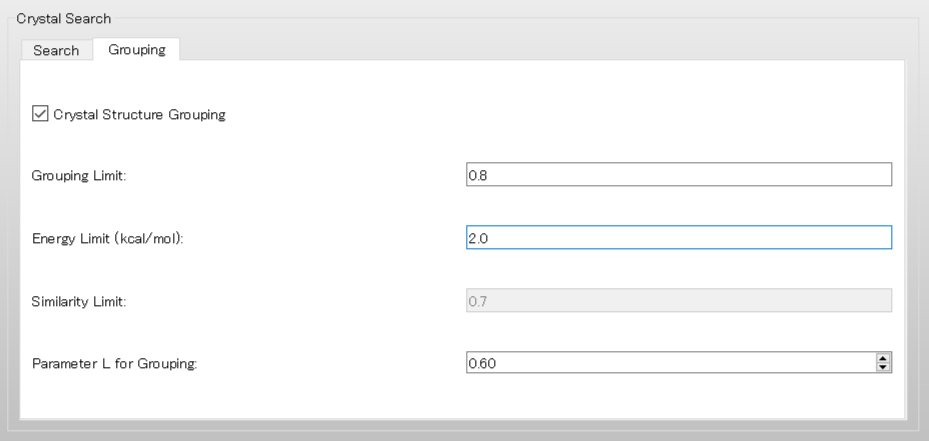
First, to group the structures, check the [Crystal Structure Grouping] checkbox and set the [Energy Limit (kcal/mol):] to 2.0.
This setting means that only structures within 2.0 kcal/mol of the lowest crystal energy will be included in the grouping process.
The [Grouping Limit:] value of 0.8 indicates that structures with a PXRD similarity value greater than 0.8 will be assigned to the same group.
The maximum similarity value is 1.0, which corresponds to a perfect match between PXRD patterns.
By adjusting the grouping limit, the degree of similarity required for structures to be grouped together can be controlled.
When completing the settings, click to start the calculation.
If you performed the initial search by manually adding keywords, you must click the and add the same keywords again when restarting.
After doing so, click . Note that if the settings of the search do not match those of the initial calculation, the restart may not function correctly.
If you want to perform both the crystal structure search and grouping in a single job without using the restart function, be sure to configure both the search and grouping settings within the [Crystal Search] dialog before submitting the job.
[If you executed the search using command line]
Store the II-c2.mol and II-c2.ini files used for the search, along with the II-c2.cpt file generated after the search, in a single folder.
Rename the extension of the II-c2.cpt file to “rst”, and add the “CSP_RESTART” keyword to the .ini file.
In addition, include the necessary keywords for grouping in the ini file.
II-c2.ini file
MMFF94S
CRYSTAL_SEARCH
CSP_SPGP=(P21/C,P-1,C2/C,P212121,P21,PBCA,PNA21,PNMA,CC,P1)
CSP_ROT_MODE=RANDOM
CSP_AUS_MODE=RANDOM
CSP_MAX_CRYSTAL=10000
CRYSTAL_OPTIMIZATION=ALL
CSP_RESTART
CSP_GROUPING=YES
CSP_GROUPING_ELIMIT=2.0
CSP_GROUPING_GLIMIT=0.8
The parts shown in red are additional keywords.
[CSP_GROUPING=YES] is the keyword for executing the grouping process.
[CSP_GROUPING_ELIMIT=2.0] indicates that structures within 2.0 kcal/mol of the lowest crystal energy will be included in the grouping process.
[CSP_GROUPING_GLIMIT=0.8] specifies that structures with a PXRD similarity value greater than 0.8 will be assigned to the same group.
The maximum similarity value is 1.0, which indicates a perfect match between the patterns.
By adjusting the grouping limit, the degree of similarity required for structures to be grouped together can be controlled.
[CSP_RESTART] is the keyword used to restart the crystal structure search.
Please do not modify the keywords that were originally used for the search calculation. If the search settings do not match, the restart may not function correctly.
When completing the calculation settings, execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par II-c2enter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
If you want to perform both the crystal structure search and grouping in a single job without using the restart function, be sure to configure both the search and grouping settings within the [Crystal Search] dialog before submitting the job. If you want to perform both the crystal structure search and grouping in a single job without using the restart function, prepare the above .ini file. However, you have to remove the [CSP_RESTART] keyword from the ini file, and the rst file is not required.
Results of grouping
In the section [*** GROUPS OF PREDICTED CRYSTAL STRUCTURES:] of the csp file, the results of grouping the structures found during the crystal structure search are output.
*** GROUPS OF PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV GID
217 18 9 -15.2940 4.7263 -20.0203 612.5302 1.3554 8.3759 18.2972 8.8552 90.0000 26.8303 90.0000 P21/C 365 4015 20.00 0 1
301 4 10 -15.2939 4.7218 -20.0156 612.7425 1.3549 8.3793 18.2092 9.2919 90.0000 25.6065 90.0000 P21/C 357 3927 20.00 0 1
2959 3616 109 -13.8613 4.6233 -18.4847 616.0985 1.3476 9.2116 4.0045 16.7017 90.0000 90.0000 90.0000 P212121 357 3927 20.00 0 1
3095 3653 110 -13.8590 4.6056 -18.4646 616.7098 1.3462 16.7316 9.2213 3.9972 90.0000 90.0000 90.0000 P212121 361 3971 20.00 1 1
3226 2222 118 -13.7981 4.7360 -18.5342 1323.1051 1.2550 8.3032 21.6640 8.3517 90.0000 118.2722 90.0000 C2/C 349 3839 20.00 0 1
2351 5291 74 -14.1961 4.6744 -18.8705 329.3236 1.2605 6.8715 8.3276 5.7551 90.0000 90.0000 90.0000 P21 339 3729 20.00 0 2
2355 5755 75 -14.1849 4.6625 -18.8474 329.2850 1.2607 6.8650 8.3211 5.7643 90.0000 90.0000 90.0000 P21 339 3729 20.00 1 2
2461 3632 81 -14.1273 4.6739 -18.8012 663.6963 1.2509 5.7731 8.3220 13.8144 90.0000 90.0000 90.0000 P212121 329 3619 20.00 0 2
2508 3628 84 -14.1173 4.6624 -18.7798 663.4689 1.2513 5.7835 8.3164 13.7941 90.0000 90.0000 90.0000 P212121 325 3575 20.00 1 2
2535 62 85 -14.1158 4.6803 -18.7961 652.6047 1.2722 8.3470 4.0014 21.6032 90.0000 115.2492 90.0000 P21/C 348 3828 20.00 0 3
2562 15 86 -14.1141 4.6708 -18.7848 652.8701 1.2717 15.5715 3.9886 16.6959 90.0000 39.0207 90.0000 P21/C 345 3795 20.00 0 3
3194 3772 115 -13.8057 4.6746 -18.4803 658.9316 1.2600 8.3420 4.0148 19.6744 90.0000 90.0000 90.0000 P212121 349 3839 20.00 0 3
3222 3753 117 -13.7995 4.6615 -18.4610 659.0917 1.2597 3.9986 19.7653 8.3393 90.0000 90.0000 90.0000 P212121 343 3773 20.00 2 3
109 3622 3 -15.6125 4.7219 -20.3344 614.2783 1.3515 4.3718 19.8181 7.0899 90.0000 90.0000 90.0000 P212121 373 4103 20.00 0 4
176 51 5 -15.5986 4.7162 -20.3147 614.9313 1.3501 4.3234 7.1069 21.9413 90.0000 114.1992 90.0000 P21/C 371 4081 20.00 0 4
193 90 6 -15.4618 4.7170 -20.1789 618.7977 1.3417 4.3746 7.0720 24.0800 90.0000 123.8359 90.0000 P21/C 374 4114 20.00 0 4
397 7257 12 -15.1231 4.7333 -19.8564 631.5499 1.3146 20.5400 4.3360 7.0912 90.0000 90.0000 90.0000 PNA21 369 4059 20.00 0 4
Each group is separated by a blank line and is assigned a unique GID. For example, the group with GID 4 contains the 3rd, 5th, 6th, and 12th structures, which indicates that these structures have similar atomic arrangements (as shown in the following figure).