Crystal structure search
Polymorphism is a common phenomenon observed in organic molecules. CONFLEX can explore possible crystal structures of an organic molecule including both packing and conformational polymorphs, using the original algorithm [Ishii, H., Obata, S., Niitsu, N. et al. Sci. Rep. 10, 2524 (2020).].
[Crystal structure search from a structural formula]
The Cambridge Crystallographic Data Centre (CCDC) regularly conducts blind tests of crystal structure prediction (CSP) [P.M. Lommerse, et al, Acta Cryst. B56, 697-714, 2000]. To explain crystal structure prediction using the CONFLEX program, we use 5-cyano-3-hydroxythiophene (II), which was used in the CSP blind test (see the figure below).
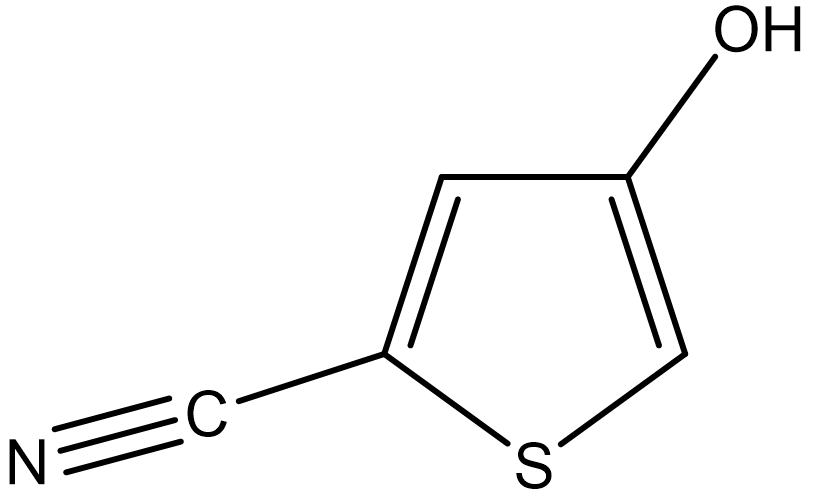
First, we create a molecular file for II using PerkinElmer ChemDraw software. For instructions on how to use ChemDraw, please refer to its user manual.
The molecular file for II is saved as “II.mol” in the MDL MOL file format.
II.mol file
II.mol
ChemDraw08301910352D
8 8 0 0 0 0 0 0 0 0999 V2000
0.2633 0.3011 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.0883 0.3011 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0083 -0.4836 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.6758 -0.9685 0.0000 S 0 0 0 0 0 0 0 0 0 0 0 0
1.3432 -0.4836 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.5732 0.9685 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.7763 -0.7385 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.5732 -0.9520 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
1 3 2 0
3 4 1 0
4 5 1 0
5 2 2 0
2 6 1 0
3 7 1 0
7 8 3 0
M END
Next, we optimize the structure of single molecule of II.
[Execution from Interface]
Open the II.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. This will be started the geometry optimization of II.
[Execution from command line]
Save the II.mol file in a folder, and then execute the following command. This will be started the geometry optimization of II. If an ini file is not provided, CONFLEX will perform the optimization using the default settings.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par IIenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Next, we perform a conformation search for II.
[Execution from Interface]
Open the II-F.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and select [Conformation Search] from the [Calculation Type:] pull-down menu on the calculation setting dialog that appears.
Edit the value of [Search Limit:] to 10.0. This parameter is used as a criterion for selecting the initial structures in the conformational search.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the II-F.ini file.
II-F.ini file
CONFLEX SEL=10.0
[CONFLEX] means that the conformation search is performed.
[SEL=10.0] means that the search limit, which is used as a criterion for selecting the initial structure in the conformation search, is set to 10.0 kcal/mol.
Store the two files of II-F.mol and II-F.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par II-Fenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
After the conformation search, we obtain two conformers of II. Here, we use the second most stable conformer for the crystal structure search calculation.
We extract the structure data of the selected conformer from the “II-F.sdf” file and save it as “II-c2.mol”. The II-F.sdf file is located in the folder containing the II-F.mol file. If you performed the conformational search using the Interface, the file name may be II-F_conv.sdf.
II-c2.mol file
II.mol
CONFLEX 19083010373D 1 1.00000 4.54816 1
CS ,E = 4.548, G = 0.974E-10, P = 48.3970, M( 0), IFN =00000002-00000001
11 11 0 0 999 V2000
1.1214 -0.0104 0.0000 C 0 0 0 0 0
0.7822 -1.3858 0.0000 C 0 0 0 0 0
-0.0000 0.7946 -0.0000 C 0 0 0 0 0
-1.4433 -0.1296 -0.0000 S 0 0 0 0 0
-0.5732 -1.6069 0.0000 C 0 0 0 0 0
1.6924 -2.3749 0.0000 O 0 0 0 0 0
0.0137 2.2245 -0.0000 C 0 0 0 0 0
0.0454 3.3854 -0.0000 N 0 0 0 0 0
2.1374 0.3665 0.0000 H 0 0 0 0 0
-1.1076 -2.5464 0.0000 H 0 0 0 0 0
1.2623 -3.2477 0.0000 H 0 0 0 0 0
1 2 1 0 0
1 3 2 0 0
3 4 1 0 0
4 5 1 0 0
5 2 2 0 0
2 6 1 0 0
3 7 1 0 0
7 8 3 0 0
1 9 1 0 0
5 10 1 0 0
6 11 1 0 0
M END
Next, we perform a crystal structure search of II.
[Execution from Interface]
Open the II-c2.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
To perform the crystal structure search, in [General Settings] dialog of the detailed settings dialog, select [Crystal Search] from the pull-down menu of [Calculation Type:].
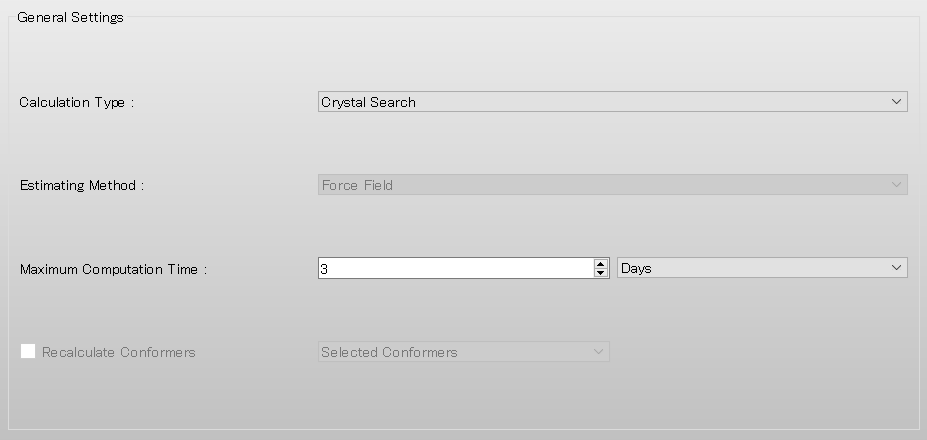
The default method for crystal structure optimization is [ALL]. You can change the optimization method by selecting a different option from the [Crystal Optimization:] pull-down menu in the [Crystal Calculation] dialog. In this dialog, you can also change settings for calculating intermolecular interactions such as the cutoff distance and the method for calculating Coulombic interactions, and other related parameters.
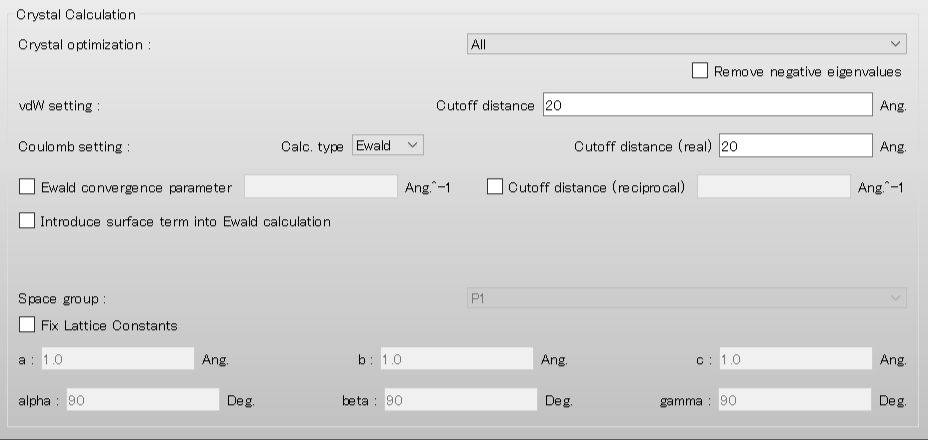
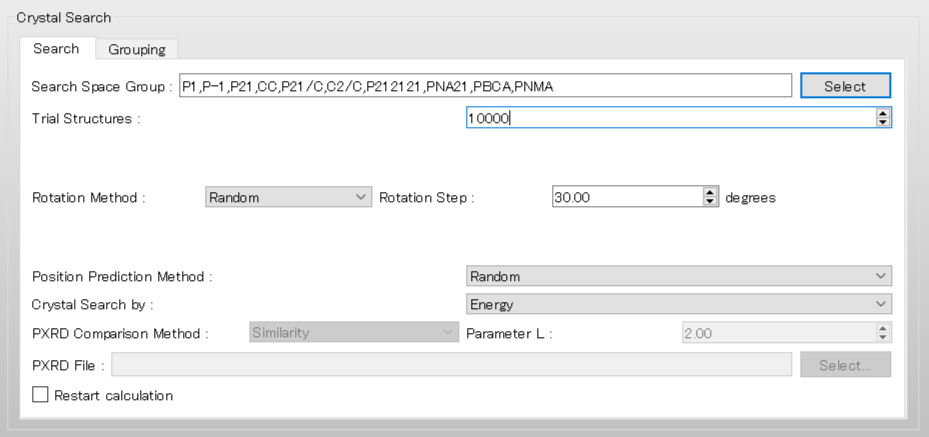
Next, we set the parameters for the crystal structure search in the [Crystal Search] dialog. First, to select the space groups using in the search, click of [Search Space Group:].
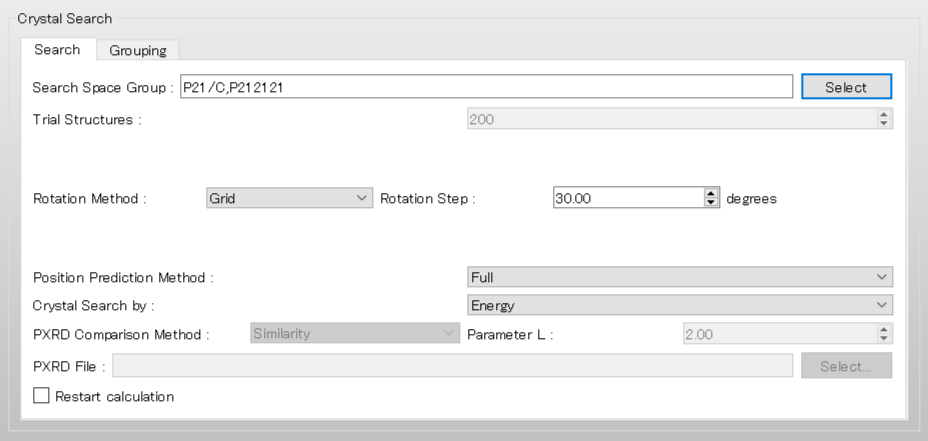
The dialog that appears displays the top 10 space groups based on the space group frequency ranking provided by the CCDC. Select all space groups by checking their checkboxes, and then click .
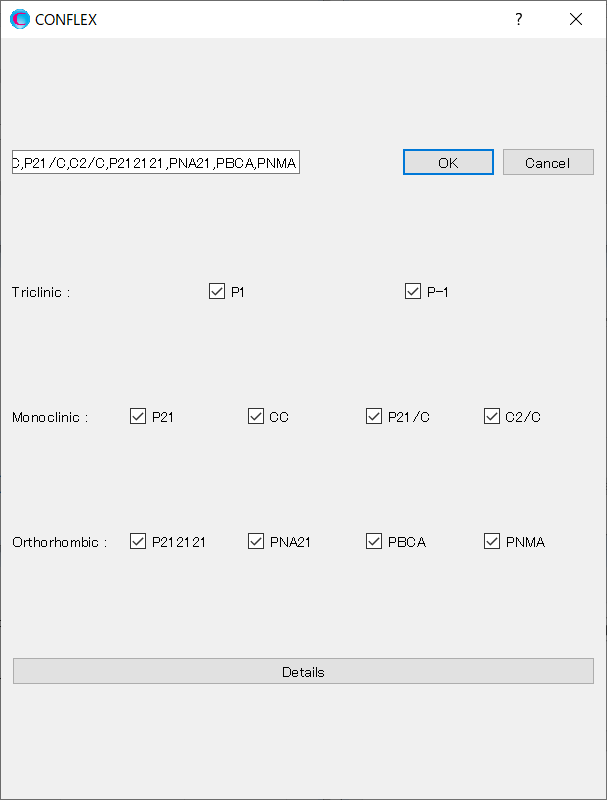
[Rotation Method:] and [Position Prediction Method:] set the methods for deteriming the initial molecular orientation and position, respectively.
Here, both methods are set to [Random]. If you want to search in detail, each method should be set to [Grid] and [Full], respectively.
[Trial Structures:] sets the number of trial structures that are created for the search. Here, the number of trial structures is set to 10000.
If you use a larger number, you can perform a more accurate search, but the computational cost will be high.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the II-c2.ini file.
II-c2.ini file
CRYSTAL_SEARCH CSP_SPGP=(P21/C,P-1,C2/C,P212121,P21,PBCA,PNA21,PNMA,CC,P1) CSP_ROT_MODE=RANDOM CSP_AUS_MODE=RANDOM CSP_MAX_CRYSTAL=10000 CRYSTAL_OPTIMIZATION=ALL
[CRYSTAL_SEARCH] means to execute a crystal structure search.
The space groups used in the search are defined by the keyword [CSP_SPGP=]. Here, we use P21/c, P-1, C2/c, P212121, P21, Pbca, Pna21, Pnma, Cc, and P1, which are the top 10 space groups based on the space group frequency ranking provided by the CCDC.
[CSP_ROT_MODE=] and [CSP_AUS_MODE=] set the methods for determining the initial molecular orientation and position, respectively.
Here, both methods are set to [Random]. If you want to search in detail, each method should be set to [Grid] and [Full], respectively.
[CSP_MAX_CRYSTAL=] sets the number of trial structures that are created for the search. Here, the number of trial structures is set to 10000.
Using a larger number will give a more accurate search, but the computational cost will be high.
The method of crystal structure optimization is defined by the [CRYSTAL_OPTIMIZATION=] keyword and is set to “ALL”.
The “ALL” optimization relaxes the molecular geometry, position, and orientation and cell dimensions of the crystal.
Store the two files of II-c2.mol and II-c2.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par II-c2enter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
After the crystal structure search is complete, you will obtain the II-C2.csp file, which contains detailed results of the search.
The [*** PREDICTED CRYSTAL STRUCTURES:] section of the II-C2.csp file lists the computationally suggested polymorphs of II in order of crystal energy.
The structural data of these polymorphs are stored in the II-C2-PCS.cif file.
*** PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV
4 318 1 -15.7921 4.6942 -20.4864 607.6657 1.3663 9.5996 8.3419 10.7545 90.0000 44.8779 90.0000 P21/C 365 4015 20.00 0
30 5295 2 -15.6634 4.7074 -20.3708 306.9963 1.3522 13.4774 7.0951 4.3469 90.0000 132.3915 90.0000 P21 371 4081 20.00 0
147 3622 3 -15.6125 4.7219 -20.3344 614.2783 1.3515 4.3718 19.8181 7.0899 90.0000 90.0000 90.0000 P212121 373 4103 20.00 0
167 3853 4 -15.6061 4.6925 -20.2986 612.8432 1.3547 9.6310 8.3434 7.6267 90.0000 90.0000 90.0000 P212121 325 3575 20.00 0
178 51 5 -15.5986 4.7162 -20.3147 614.9313 1.3501 4.3234 7.1069 21.9413 90.0000 114.1992 90.0000 P21/C 371 4081 20.00 0
190 90 6 -15.4618 4.7170 -20.1789 618.7977 1.3417 4.3746 7.0720 24.0800 90.0000 123.8359 90.0000 P21/C 374 4114 20.00 0
195 1948 7 -15.3029 4.7145 -20.0173 1222.4436 1.3583 14.5999 8.3633 10.4384 90.0000 73.5604 90.0000 C2/C 357 3927 20.00 0
206 169 8 -15.2948 4.7210 -20.0158 612.9536 1.3545 4.0230 8.3785 19.4984 90.0000 111.1490 90.0000 P21/C 358 3938 20.00 0
229 18 9 -15.2940 4.7263 -20.0203 612.5302 1.3554 8.3759 18.2972 8.8552 90.0000 26.8303 90.0000 P21/C 365 4015 20.00 0
290 4 10 -15.2939 4.7218 -20.0156 612.7425 1.3549 8.3793 18.2092 9.2919 90.0000 25.6065 90.0000 P21/C 357 3927 20.00 0
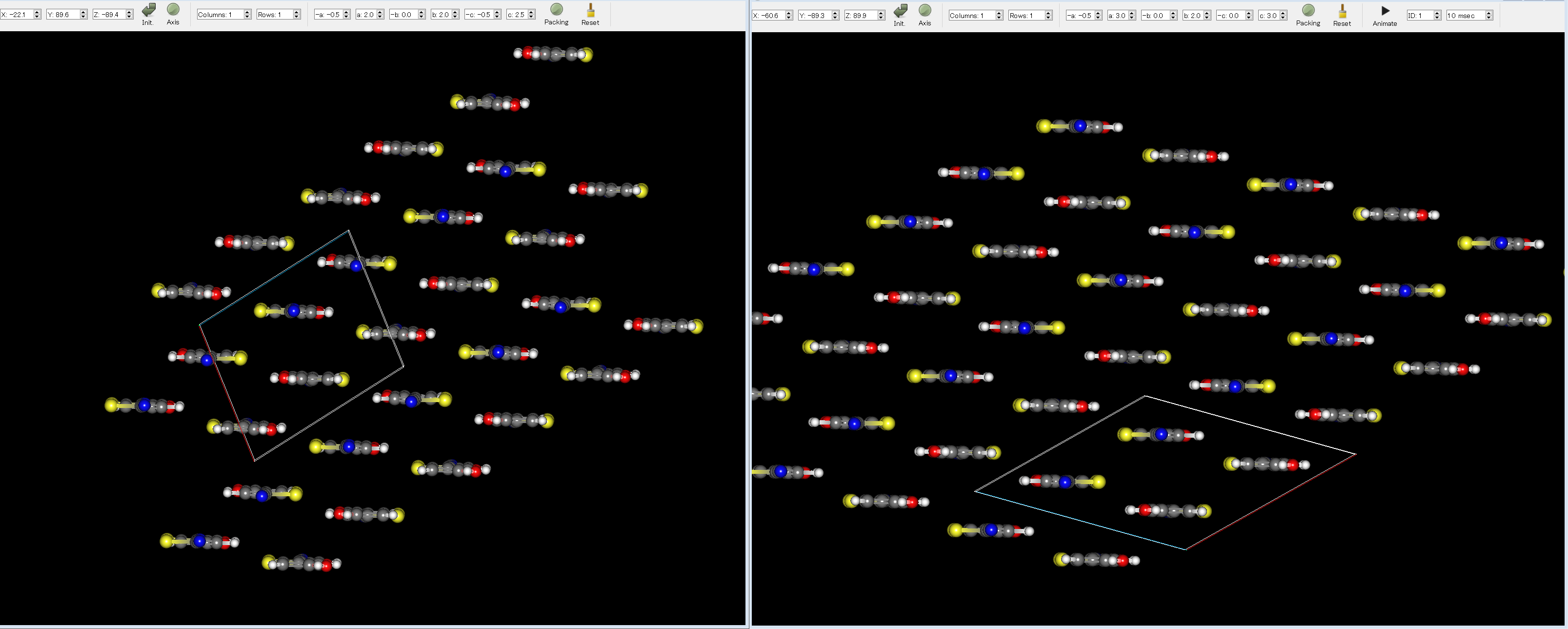
Here, by comparing the experimental structure (left figure) with the first structure (right figure), you can see that the two structures match well.
[Crystal structure search for a multi-component system]
This section describes a crystal structure search for an asymmetric unit containing multiple components.
Here, we use a co-crystal of 2-amino-4-methylpyrimidine and 2-methylbenzoic acid, which was employed in the CSP blind test.
First, molecular structures of 2-amino-4-methylpyrimidine and 2-methylbenzoic acid are created using PerkinElmer ChemDraw software.
Please refer to the ChemDraw manual for instructions on how to use the software.
The molecular file of 2-amino-4-methylpyrimidine is saved as “AMP.mol” in the MDL MOL file format.
The molecular file of 2-amino-4-methylpyrimidine is saved in MDL MOL format as “AMP.mol”.
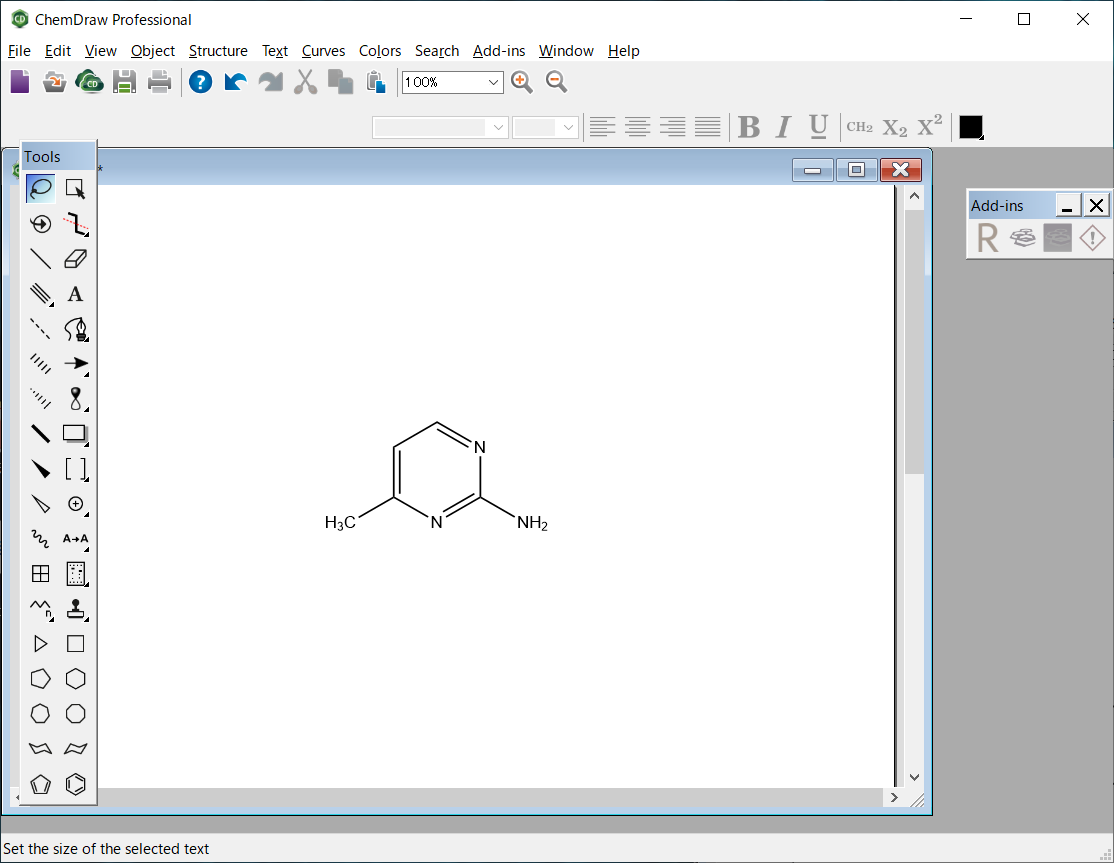
AMP.mol file
AMP.mol
ChemDraw09022015212D
8 8 0 0 0 0 0 0 0 0999 V2000
-0.7145 0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.7145 -0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0000 -0.8250 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
0.7145 -0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7145 0.4125 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.0000 0.8250 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.4289 -0.8250 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4289 -0.8250 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0
2 3 1 0
3 4 2 0
4 5 1 0
5 6 2 0
6 1 1 0
2 7 1 0
4 8 1 0
M END
The molecular file of 2-methylbenzoic acid is saved as “MBA.mol” in the MDL MOL file format.
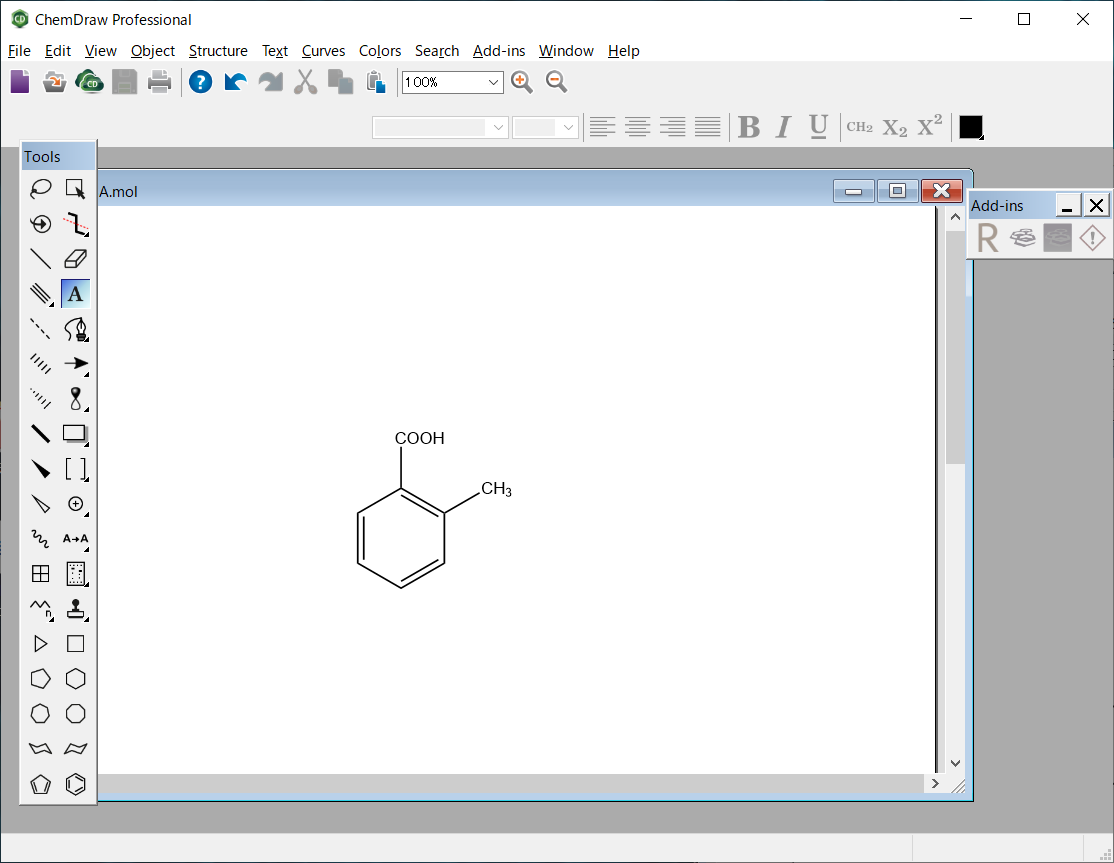
MBA.mol file
MBA.mol
ChemDraw09022015232D
10 10 0 0 0 0 0 0 0 0999 V2000
-1.0717 -0.2062 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.0717 -1.0313 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3572 -1.4438 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.3572 -1.0313 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.3572 -0.2062 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3572 0.2062 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.0717 0.2062 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3572 1.0313 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.0717 1.4438 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.3572 1.4438 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
8 9 2 0
8 10 1 0
6 8 1 0
1 2 2 0
2 3 1 0
3 4 2 0
4 5 1 0
5 6 2 0
6 1 1 0
5 7 1 0
M STY 1 1 SUP
M SLB 1 1 1
M SAL 1 3 8 9 10
M SBL 1 1 3
M SMT 1 COOH
M SBV 1 3 0.0000 -0.8250
M END
Next, geometry optimizations of 2-amino-4-methylpyrimidine and 2-methylbenzoic acid are performed.
[Execution from Interface]
Open the AMP.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. The geometry optimization of 2-amino-4-methylpyrimidine will be started.
Next, open the MBA.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. The geometry optimization of 2-methylbenzoic acid will be started.
[Execution from command line]
Store the AMP.mol and MBA.mol in a folder, and execute the following commands. The geometry optimizations will be started.
When ini files are not prepared, CONFLEX carries out the optimizations of input structures with the default settings.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par AMPenterC:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par MBAenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
The 2-methylbenzoic acid molecule has several conformations. Therefore, a conformation search should be performed.
[Execution from Interface]
Open the MBA-F.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and select [Conformation Search] from the [Calculation Type:] pull-down menu on the calculation setting dialog that appears.
Edit the value of [Search Limit:] to 10.0. This parameter is used as a criterion for selecting the initial structures in the conformation search.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the MBA-F.ini file.
MBA-F.ini file
CONFLEX SEL=10.0
[CONFLEX] means that the conformation search is performed.
[SEL=10.0] means that the search limit, which is used as a criterion for selecting the initial structure in the conformation search, is set to 10.0 kcal/mol.
Store the two files of MBA-F.mol and MBA-F.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par MBA-Fenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
After the conformational search, seven conformers of 2-methylbenzoic acid are obtained.
Here, we use the most stable conformer for the next calculation. The structural data of the selected conformer is extracted from “MBA-F.sdf” file and saved as “MBA-c1.mol”.
The “MBA-F.sdf” file is located in the same folder as the “MBA-F.mol” file.
MBA-c1.mol file
MBA.mol
CONFLEX 20090215293D 1 1.00000 16.90241 3
CS ,E = 16.902, G = 0.213E-06, P = 89.7913, M( 0), IFN =00000001-00000003
18 18 0 0 999 V2000
-1.3006 0.8083 0.0000 C 0 0 0 0 0
-2.4209 -0.0240 0.0000 C 0 0 0 0 0
-2.2574 -1.4041 0.0000 C 0 0 0 0 0
-0.9754 -1.9534 -0.0000 C 0 0 0 0 0
0.1671 -1.1315 -0.0000 C 0 0 0 0 0
-0.0000 0.2708 0.0000 C 0 0 0 0 0
1.5196 -1.7909 -0.0000 C 0 0 0 0 0
1.1760 1.1942 0.0000 C 0 0 0 0 0
2.3544 0.8930 0.0000 O 0 0 0 0 0
0.8165 2.4930 -0.0000 O 0 0 0 0 0
-1.4568 1.8849 0.0000 H 0 0 0 0 0
-3.4187 0.4075 0.0000 H 0 0 0 0 0
-3.1277 -2.0558 0.0000 H 0 0 0 0 0
-0.8717 -3.0372 -0.0000 H 0 0 0 0 0
2.0811 -1.5139 0.8977 H 0 0 0 0 0
1.4333 -2.8831 -0.0000 H 0 0 0 0 0
2.0811 -1.5139 -0.8977 H 0 0 0 0 0
1.6744 2.9669 -0.0000 H 0 0 0 0 0
8 9 2 0 0
8 10 1 0 0
6 8 1 0 0
1 2 2 0 0
2 3 1 0 0
3 4 2 0 0
4 5 1 0 0
5 6 2 0 0
6 1 1 0 0
5 7 1 0 0
1 11 1 0 0
2 12 1 0 0
3 13 1 0 0
4 14 1 0 0
7 15 1 0 0
7 16 1 0 0
7 17 1 0 0
10 18 1 0 0
M END
Next, using the AMP-F.mol and MBA-c1.mol files, a structure file for the molecular complex of 2-amino-4-methylpyrimidine and 2-methylbenzoic acid is created and saved as “XV.mol”.
The XV.mol file is generated by adding the atomic coordinates and bond information from the MBA-c1.mol file to the AMP-F.mol file.
The 4th line in the XV.mol file, “33 33”, represents the number of atoms and bonds in the complex, respectively.
The bond information, “ 1 2 2 0 0”, indicates that atoms 1 and 2 are bonded by a double bond. Therefore, when adding the bond information of 2-methylbenzoic acid to the AMP-F.mol file, you should update the original atom serial numbers in the bond information. For example, change “ 8 9 2 0 0” to “ 23 24 2 0 0”.
XV.mol file
XV.mol
33 33 0 0 999 V2000
1.2096 -1.0822 -0.0000 C 0 0 0 0 0
1.1638 0.2958 0.0000 C 0 0 0 0 0
-0.0000 0.9788 0.0000 N 0 0 0 0 0
-1.1295 0.2628 0.0000 C 0 0 0 0 0
-1.1861 -1.0719 0.0000 N 0 0 0 0 0
-0.0075 -1.7194 -0.0000 C 0 0 0 0 0
2.4311 1.0955 -0.0000 C 0 0 0 0 0
-2.3091 0.9449 -0.0000 N 0 0 0 0 0
2.1397 -1.6334 -0.0000 H 0 0 0 0 0
-0.0748 -2.8030 -0.0000 H 0 0 0 0 0
2.2152 2.1688 0.0000 H 0 0 0 0 0
3.0226 0.8673 0.8921 H 0 0 0 0 0
3.0226 0.8673 -0.8921 H 0 0 0 0 0
-3.1598 0.4121 -0.0000 H 0 0 0 0 0
-2.2706 1.9481 -0.0000 H 0 0 0 0 0
-1.3006 0.8083 0.0000 C 0 0 0 0 0
-2.4209 -0.0240 0.0000 C 0 0 0 0 0
-2.2574 -1.4041 0.0000 C 0 0 0 0 0
-0.9754 -1.9534 -0.0000 C 0 0 0 0 0
0.1671 -1.1315 -0.0000 C 0 0 0 0 0
-0.0000 0.2708 0.0000 C 0 0 0 0 0
1.5196 -1.7909 -0.0000 C 0 0 0 0 0
1.1760 1.1942 0.0000 C 0 0 0 0 0
2.3544 0.8930 0.0000 O 0 0 0 0 0
0.8165 2.4930 -0.0000 O 0 0 0 0 0
-1.4568 1.8849 0.0000 H 0 0 0 0 0
-3.4187 0.4075 0.0000 H 0 0 0 0 0
-3.1277 -2.0558 0.0000 H 0 0 0 0 0
-0.8717 -3.0372 -0.0000 H 0 0 0 0 0
2.0811 -1.5139 0.8977 H 0 0 0 0 0
1.4333 -2.8831 -0.0000 H 0 0 0 0 0
2.0811 -1.5139 -0.8977 H 0 0 0 0 0
1.6744 2.9669 -0.0000 H 0 0 0 0 0
1 2 2 0 0
2 3 1 0 0
3 4 2 0 0
4 5 1 0 0
5 6 2 0 0
6 1 1 0 0
2 7 1 0 0
4 8 1 0 0
1 9 1 0 0
6 10 1 0 0
7 11 1 0 0
7 12 1 0 0
7 13 1 0 0
8 14 1 0 0
8 15 1 0 0
23 24 2 0 0
23 25 1 0 0
21 23 1 0 0
16 17 2 0 0
17 18 1 0 0
18 19 2 0 0
19 20 1 0 0
20 21 2 0 0
21 16 1 0 0
20 22 1 0 0
16 26 1 0 0
17 27 1 0 0
18 28 1 0 0
19 29 1 0 0
22 30 1 0 0
22 31 1 0 0
22 32 1 0 0
25 33 1 0 0
M END
Next, to determine the possible molecular positions of 2-amino-4-methylpyrimidine and 2-methylbenzoic acid in the molecular complex, a host-ligand configuration search is performed.
[Execution from Interface]
Open the XV.mol file using CONFLEX Interface. Initially, the two molecules are in a collision, but their positions are changed according to a search algorithm.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
To perform the host-ligand configuration search, in [General Settings] dialog of the detailed settings dialog, select [Host Ligand Search] from the pull-down menu of [Calculation Type:].
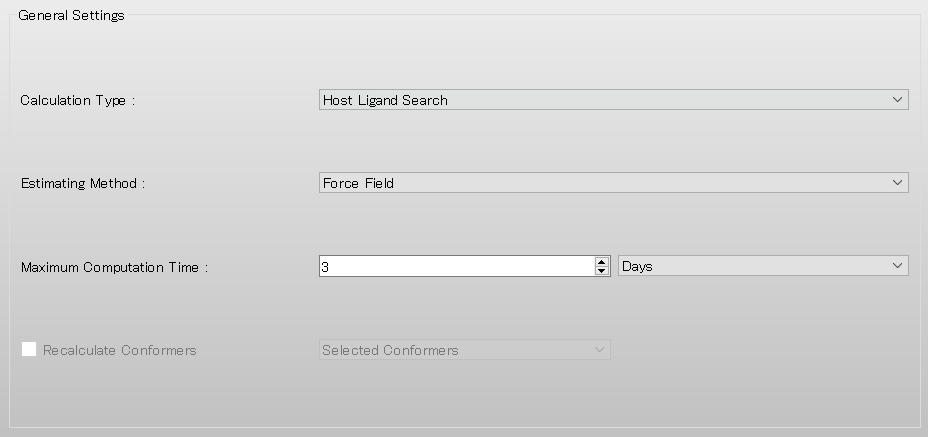
The settings for host-ligand configuration search calculation are configured in the [Host Ligand Search] dialog.
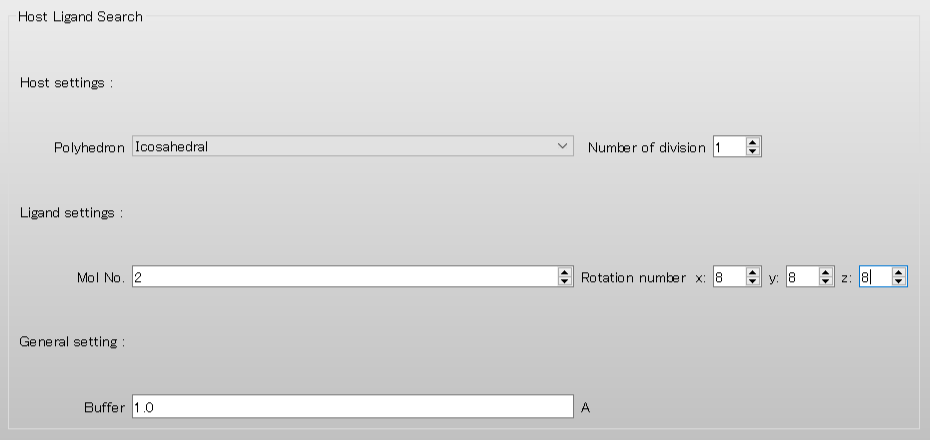
To rotate the 2-methylbenzoic acid molecule, assigned as the ligand, by 45 degrees in each step, set the values of “Rotational number x, y, z” to 8, respectively. When completing the settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the XV.ini file.
XV.ini file
HLSEARCH HLSEARCH_LIGAND_ROT=(8,8,8)
[HLSEARCH] means to execute a host-ligand configuration search calculation.
[HLSEARCH_LIGAND_ROT=(8,8,8)] is used to rotate the 2-methylbenzoic acid molecule, assigned as the ligand, by 45 degrees in each step around x, y, and z axes.
Store the two files of XV.mol and XV.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par XVenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
After the calculation, 47 structures of the complex are obtained. The most stable structure is then selected for the crystal structure search. The structural data of the selected structure is extracted from the “XV.sdf” file and saved as “XV-c1.mol”. The XV.sdf file is located in the same folder as the XV.mol file.
XV-c1.mol file
XV.mol
CONFLEX 20090215503D 1 1.00000 -86.91814 17
NONE,E = -86.918, G = 0.249E-06, P = 20.9930, M( 0), IFN =00000001-00000017
33 33 0 0 999 V2000
-4.6389 1.0672 0.0877 C 0 0 0 0 0
-3.2840 1.2059 0.3058 C 0 0 0 0 0
-2.4142 0.1779 0.1589 N 0 0 0 0 0
-2.9370 -1.0122 -0.1834 C 0 0 0 0 0
-4.2342 -1.2388 -0.4067 N 0 0 0 0 0
-5.0617 -0.1892 -0.2687 C 0 0 0 0 0
-2.7235 2.5426 0.6933 C 0 0 0 0 0
-2.0889 -2.0671 -0.3182 N 0 0 0 0 0
-5.3328 1.8902 0.1921 H 0 0 0 0 0
-6.1102 -0.3980 -0.4587 H 0 0 0 0 0
-1.6892 2.4500 1.0374 H 0 0 0 0 0
-2.7441 3.2203 -0.1656 H 0 0 0 0 0
-3.3069 2.9800 1.5099 H 0 0 0 0 0
-2.4812 -2.9552 -0.5738 H 0 0 0 0 0
-1.0952 -1.9477 -0.1563 H 0 0 0 0 0
2.8428 1.4208 -0.4637 C 0 0 0 0 0
4.1800 1.8122 -0.5390 C 0 0 0 0 0
5.1849 0.9050 -0.2233 C 0 0 0 0 0
4.8550 -0.3923 0.1698 C 0 0 0 0 0
3.5126 -0.8046 0.2572 C 0 0 0 0 0
2.4937 0.1165 -0.0682 C 0 0 0 0 0
3.2239 -2.2160 0.6905 C 0 0 0 0 0
1.0518 -0.2696 -0.0173 C 0 0 0 0 0
0.6010 -1.3998 0.0329 O 0 0 0 0 0
0.2426 0.8063 -0.0191 O 0 0 0 0 0
2.0730 2.1442 -0.7236 H 0 0 0 0 0
4.4339 2.8231 -0.8471 H 0 0 0 0 0
6.2281 1.2054 -0.2833 H 0 0 0 0 0
5.6587 -1.0857 0.4113 H 0 0 0 0 0
2.7876 -2.7870 -0.1349 H 0 0 0 0 0
4.1382 -2.7349 0.9990 H 0 0 0 0 0
2.5491 -2.2242 1.5524 H 0 0 0 0 0
-0.6842 0.4546 0.0376 H 0 0 0 0 0
1 2 2 0 0
2 3 1 0 0
3 4 2 0 0
4 5 1 0 0
5 6 2 0 0
6 1 1 0 0
2 7 1 0 0
4 8 1 0 0
1 9 1 0 0
6 10 1 0 0
7 11 1 0 0
7 12 1 0 0
7 13 1 0 0
8 14 1 0 0
8 15 1 0 0
23 24 2 0 0
23 25 1 0 0
21 23 1 0 0
16 17 2 0 0
17 18 1 0 0
18 19 2 0 0
19 20 1 0 0
20 21 2 0 0
21 16 1 0 0
20 22 1 0 0
16 26 1 0 0
17 27 1 0 0
18 28 1 0 0
19 29 1 0 0
22 30 1 0 0
22 31 1 0 0
22 32 1 0 0
25 33 1 0 0
M END
Finally, we carry out the crystal structure search.
[Execution from Interface]
Open the XV-c1.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
To perform the crystal structure search, in [General Settings] dialog of the detailed settings dialog, select [Crystal Search] from the pull-down menu of [Calculation Type:].
The method of crystal structure optimization is [ALL] by default. You can change the type of crystal structure optimization by the pull-down menu of [Crystal optimization:] in [Crystal Calculation] dialog.
In this dialog, you can also change settings for calculating intermolecular interactions such as the cutoff distance and the method for calculating Coulombic interactions, and other related parameters.
Next, we configure the settings for the crystal structure search using the [Crystal Search] dialog.
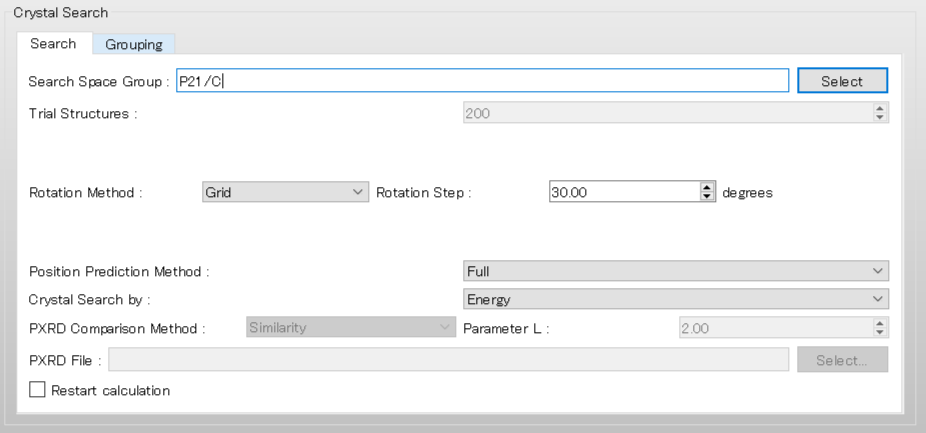
To use the space group of P21/c, edit the [Search Space Group:] from “P21/C,P212121” to “P21/C”. Also set the [Rotation Step:] value to 20.00 to rotate the molecular complex in 20-degree increments.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the XV-c1.ini file.
XV-c1.ini file
CRYSTAL_SEARCH CSP_SPGP=(P21/C) CSP_RSTEP=20.0
[CRYSTAL_SEARCH] means to execute the crystal structure search.
The space groups used in the crystal structure search are defined by “CSP_SPGP=” keyword. Here, “CSP_SPGP=(P21/C)” is set, meaning that the P21/c is applied to the search.
Store the two files of XV-c1.mol and XV-c1.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par XV-c1enter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
After the calculation, the XV-c1.csp file is generated, which contains detailed results of the search.
The [*** PREDICTED CRYSTAL STRUCTURES:] section of the XV-c1.csp file lists the computationally suggested polymorphs of 2-amino-4-methylpyrimidine and 2-methylbenzoic acid co-crystal in order of crystal energy.
the structure data of the polymorphs are stored in the XV-c1-PCS.cif file.
*** PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV
46 19 1 -112.2561 -73.1657 -39.0905 1384.6750 1.1758 6.3597 14.7215 17.2181 90.0000 120.8001 90.0000 P21/C 426 7035 20.00 0
54 487 2 -112.1447 -72.8870 -39.2577 1330.3934 1.2238 7.4330 26.1512 7.7427 90.0000 62.1242 90.0000 P21/C 436 7188 20.00 0
55 6106 3 -112.1190 -73.0217 -39.0973 1373.5185 1.1854 7.2525 14.8932 15.7678 90.0000 53.7521 90.0000 P21/C 421 6951 20.00 0
57 1692 4 -112.0257 -72.0386 -39.9871 1368.6700 1.1895 8.9646 9.9375 27.1501 90.0000 145.5370 90.0000 P21/C 431 7128 20.00 0
66 3542 5 -111.8336 -73.1270 -38.7065 1409.7885 1.1549 13.5655 12.0776 27.5867 90.0000 18.1748 90.0000 P21/C 421 6948 20.00 0
81 1208 6 -111.7751 -73.3963 -38.3788 1415.1006 1.1505 13.0894 10.0679 11.9525 90.0000 63.9490 90.0000 P21/C 430 7092 20.00 0
109 33 7 -111.7587 -73.2931 -38.4656 1403.7919 1.1598 26.0879 11.1575 8.6903 90.0000 146.2923 90.0000 P21/C 420 6936 20.00 0
134 4315 8 -111.7046 -72.2275 -39.4771 1399.2931 1.1635 7.1345 12.0828 25.4413 90.0000 39.6451 90.0000 P21/C 426 7035 20.00 0
140 1769 9 -111.5333 -72.9261 -38.6073 1414.9373 1.1507 12.3562 8.0869 14.2635 90.0000 96.8972 90.0000 P21/C 412 6807 20.00 0
141 967 10 -111.5221 -72.4012 -39.1210 1351.3577 1.2048 9.9354 10.2689 13.7854 90.0000 106.0926 90.0000 P21/C 432 7152 20.00 0
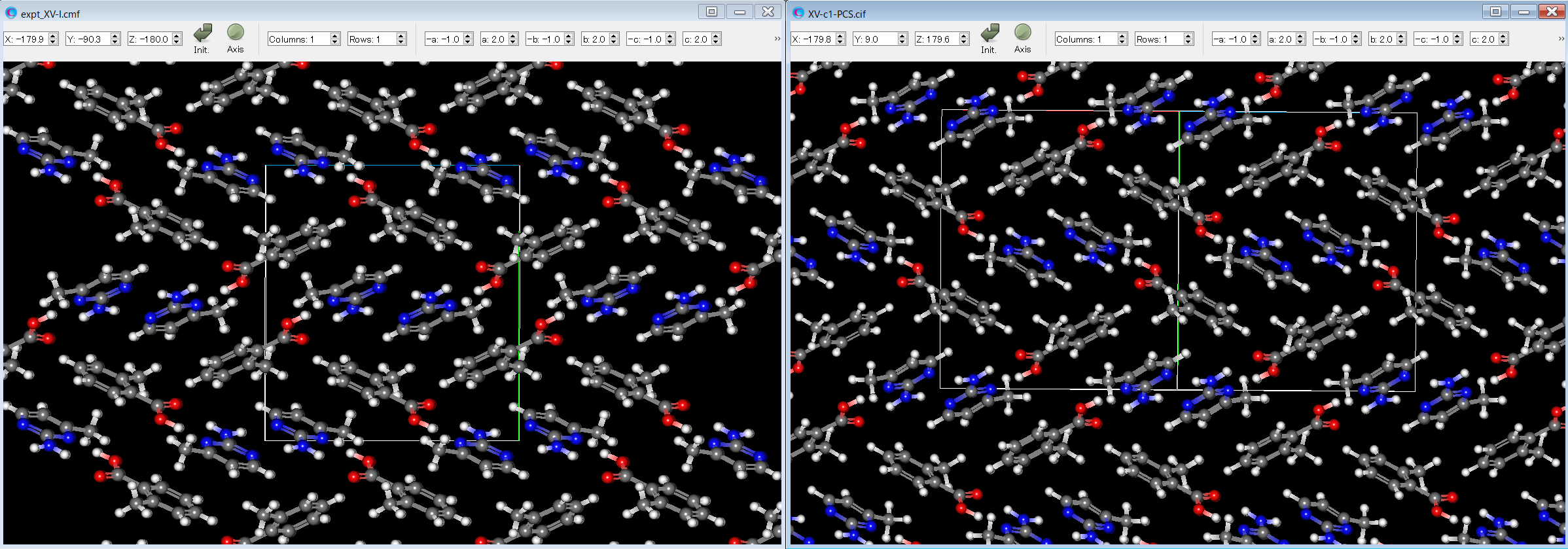
Here, by comparing the experimental structure (left figure) with the third structure (right figure), it can be seen that both structures match well.
[Crystal structure search using PXRD data]
We explain how to apply CONFLEX to crystal structure determination from powder X-ray diffraction (PXRD) data. In this example, we use the crystal structure of pamoic acid reported by Haynes et al. [D. A. Haynes et al., Acta Cryst. 2006, E62, o1170.]. First, a molecular file for pamoic acid is created using PerkinElmer ChemDraw software. Please refer to the ChemDraw manual for instructions on how to use the software. The molecular file of pamoic acid is saved as “pamoic-acid.mol” in MDL MOL file format.
pamoic-acid.mol file
pamoic-acid.mol
ChemDraw10031813162D
29 32 0 0 0 0 0 0 0 0999 V2000
-2.1471 -0.8250 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.4326 -0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.4326 0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.1471 0.8250 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8616 0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8616 -0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5761 -0.8250 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5761 -1.6500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8616 -2.0625 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.1471 -1.6500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.4294 -1.0931 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7182 -0.4538 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4326 -0.8662 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1471 -0.4538 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1471 0.3712 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4326 0.7837 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7182 0.3712 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4326 1.6087 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7182 2.0212 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0037 1.6087 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0037 0.7837 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4326 -1.6912 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
2.8616 -0.8663 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
3.5761 -0.4538 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
2.8616 -1.6913 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-2.1471 1.6500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8616 2.0625 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-1.4326 2.0625 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.7182 0.8250 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0
2 3 1 0
3 4 2 0
4 5 1 0
5 6 2 0
6 1 1 0
6 7 1 0
7 8 2 0
8 9 1 0
9 10 2 0
10 1 1 0
2 11 1 0
11 12 1 0
12 13 1 0
13 14 2 0
14 15 1 0
15 16 2 0
16 17 1 0
17 12 2 0
16 18 1 0
18 19 2 0
19 20 1 0
20 21 2 0
21 17 1 0
13 22 1 0
14 23 1 0
23 24 2 0
23 25 1 0
4 26 1 0
26 27 2 0
26 28 1 0
3 29 1 0
M END
Next, we perform geometry optimization of the single molecule of pamoic acid.
[Execution from Interface]
Open the pamoic-acid.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. The geometry optimization of II will be started.
[Execution from command line]
Store the pamoic-acid.mol in a folder, and execute the following command. The geometry optimization of pamoic acid will be started. When ini files are not prepared, CONFLEX carries out the optimization of input structure with the default settings.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par pamoic-acidenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Next, we perform a conformation search of pamoic acid.
[Execution from Interface]
Open the pamoic-acid-F.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and select [Conformation Search] from the [Calculation Type:] pull-down menu on the calculation setting dialog that appears.
Edit the value of [Search Limit:] to 30.0. This parameter is used as a criterion for selecting the initial structures in the conformation search.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the pamoic-acid-F.ini file.
pamoic-acid-F.ini file
CONFLEX SEL=30.0
[CONFLEX] means that the conformation search is performed.
[SEL=30.0] means that the search limit, which is used as a criterion for selecting the initial structure in the conformation search, is set to 30.0 kcal/mol.
Store the two files of pamoic-acid-F.mol and pamoic-acid-F.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par pamoic-acid-Fenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
After the conformation search, 118 conformers of pamoic acid are obtained. Here, the third most stable conformer is selected for the crystal structure search calculation. The structural data of the selected conformer is extracted from “pamoic-acid-F.sdf” file and saved as “pamoic-acid-c3.mol”. The “pamoic-acid-F.sdf” file is located in the same folder as the “pamoic-acid-F.mol”.
pamoic-acid-c3.mol file
pamoic-acid.mol
CONFLEX 18102311013D 1 1.00000 57.28062 16
C2 ,E = 57.281, G = 0.144E-07, P = 4.5793, M( 0), IFN =00000003-00000016
45 48 0 0 999 V2000
-1.3167 2.0796 -0.8640 C 0 0 0 0 0
-0.1166 1.2932 -0.8876 C 0 0 0 0 0
1.0079 1.7433 -0.1606 C 0 0 0 0 0
0.9563 2.9081 0.6175 C 0 0 0 0 0
-0.2214 3.6554 0.6552 C 0 0 0 0 0
-1.3466 3.2641 -0.0791 C 0 0 0 0 0
-2.5025 4.0623 -0.0282 C 0 0 0 0 0
-3.6457 3.7218 -0.7444 C 0 0 0 0 0
-3.6457 2.5743 -1.5200 C 0 0 0 0 0
-2.5025 1.7708 -1.5777 C 0 0 0 0 0
-0.0000 0.0000 -1.6979 C 0 0 0 0 0
0.1166 -1.2932 -0.8876 C 0 0 0 0 0
-1.0079 -1.7433 -0.1606 C 0 0 0 0 0
-0.9563 -2.9081 0.6175 C 0 0 0 0 0
0.2214 -3.6554 0.6552 C 0 0 0 0 0
1.3466 -3.2641 -0.0791 C 0 0 0 0 0
1.3167 -2.0796 -0.8640 C 0 0 0 0 0
2.5025 -4.0623 -0.0282 C 0 0 0 0 0
3.6457 -3.7218 -0.7444 C 0 0 0 0 0
3.6457 -2.5743 -1.5200 C 0 0 0 0 0
2.5025 -1.7708 -1.5777 C 0 0 0 0 0
-2.1663 -1.0131 -0.2688 O 0 0 0 0 0
-2.1487 -3.3401 1.3832 C 0 0 0 0 0
-3.2090 -2.7368 1.4055 O 0 0 0 0 0
-1.9746 -4.4795 2.0733 O 0 0 0 0 0
2.1487 3.3401 1.3832 C 0 0 0 0 0
3.2090 2.7368 1.4055 O 0 0 0 0 0
1.9746 4.4795 2.0733 O 0 0 0 0 0
2.1663 1.0131 -0.2688 O 0 0 0 0 0
-0.2634 4.5620 1.2557 H 0 0 0 0 0
-2.5220 4.9676 0.5759 H 0 0 0 0 0
-4.5311 4.3490 -0.6932 H 0 0 0 0 0
-4.5334 2.2918 -2.0797 H 0 0 0 0 0
-2.5709 0.8759 -2.1893 H 0 0 0 0 0
-0.8381 -0.1310 -2.3887 H 0 0 0 0 0
0.8381 0.1310 -2.3887 H 0 0 0 0 0
0.2634 -4.5620 1.2557 H 0 0 0 0 0
2.5220 -4.9676 0.5759 H 0 0 0 0 0
4.5311 -4.3490 -0.6932 H 0 0 0 0 0
4.5334 -2.2918 -2.0797 H 0 0 0 0 0
2.5709 -0.8759 -2.1893 H 0 0 0 0 0
-2.8489 -1.3798 0.3330 H 0 0 0 0 0
-2.8335 -4.6307 2.5208 H 0 0 0 0 0
2.8335 4.6307 2.5208 H 0 0 0 0 0
2.8489 1.3798 0.3330 H 0 0 0 0 0
1 2 2 0 0
2 3 1 0 0
3 4 2 0 0
4 5 1 0 0
5 6 2 0 0
6 1 1 0 0
6 7 1 0 0
7 8 2 0 0
8 9 1 0 0
9 10 2 0 0
10 1 1 0 0
2 11 1 0 0
11 12 1 0 0
12 13 1 0 0
13 14 2 0 0
14 15 1 0 0
15 16 2 0 0
16 17 1 0 0
17 12 2 0 0
16 18 1 0 0
18 19 2 0 0
19 20 1 0 0
20 21 2 0 0
21 17 1 0 0
13 22 1 0 0
14 23 1 0 0
23 24 2 0 0
23 25 1 0 0
4 26 1 0 0
26 27 2 0 0
26 28 1 0 0
3 29 1 0 0
5 30 1 0 0
7 31 1 0 0
8 32 1 0 0
9 33 1 0 0
10 34 1 0 0
11 35 1 0 0
11 36 1 0 0
15 37 1 0 0
18 38 1 0 0
19 39 1 0 0
20 40 1 0 0
21 41 1 0 0
22 42 1 0 0
25 43 1 0 0
28 44 1 0 0
29 45 1 0 0
M END
Next, we prepare a diffraction data file for the CONFLEX program from the raw data obtained by the PXRD experiment. Haynes et al. provide the raw data for the pamoic acid crystal. Here, we use “cv6632Isup2.rtv” to create a reference diffraction data file, pamoic-acid-c3.xrd, as shown below.
pamoic-acid-c3.xrd file
Co 8.010 79.990 0.01 7199 8.010 0.000000 8.020 0.000000 8.030 0.000000 8.040 91.252560 8.050 9.827270 8.060 0.000000 (中略) 79.970 43.877350 79.980 17.738100 79.990 42.620530
The first row indicates the type of X-ray source, and the second row shows the range and step of 2-theta, as well as the total number of diffraction data points. In the first row, you can also set the wavelength of the X-ray source, rather than the atomic name characters. The diffraction data are shown in the third row and subsequent rows. The intensity data at each 2-theta point are generated by subtracting the "_pd_proc_intensity_bkg_calc" value from the "_pd_meas_counts_total" value. If the resulting intensity is negative due to the subtraction, it is set to “zero” for that point. You can find the pamoic-acid-c3.xrd file in the Sample_Files folder located in the directory where CONFLEX is installed (Sample_Files\CONFLEX\crystal\powder\pamoic-acid-c3.xrd). Do not include blank lines in the xrd file, and make sure to remove the background from the raw data before using it in CONFLEX.
Finally, we perform a crystal structure search for pamoic acid
[Execution from Interface]
Open the pamoic-acid-c3.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
To perform the crystal structure search, in [General Settings] dialog of the detailed settings dialog, select [Crystal Search] from the pull-down menu of [Calculation Type:].
The method of crystal structure optimization is [ALL] by default. You can change the type of crystal structure optimization by the pull-down menu of [Crystal optimization:] in [Crystal Calculation] dialog.
In this dialog, you can also change settings for calculating intermolecular interactions such as the cutoff distance and the method for calculating Coulombic interactions, and other related parameters.
Next, we configure the parameters for the crystal structure search using the [Crystal Search] dialog.
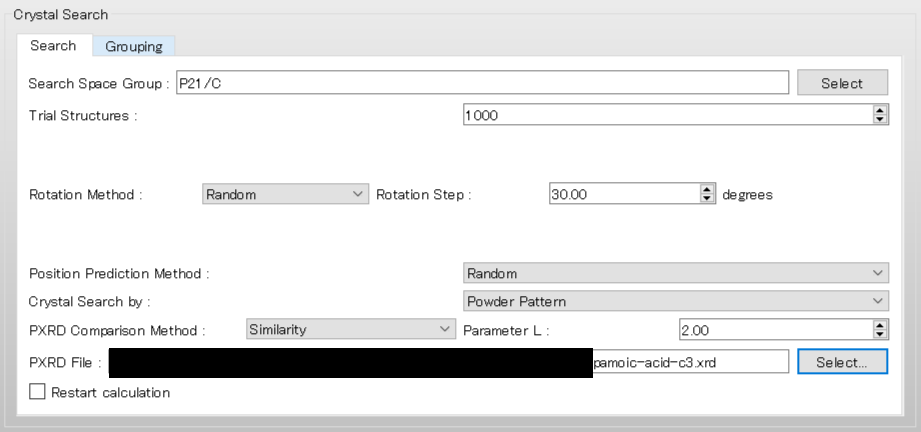
To use the space group P21/c, edit the [Search Space Group:] field from “P21/C,P212121” to “P21/C”. The [Rotation Method:] and [Position Prediction Method:] are used to determine the initial molecular orientation and position, respectively. Here, both methods are set to [Random]. The [Trial Structures:] defines the number of trial structures created for the search. Here, the number of trial structures is set to 1000.
Next, change the pull-down menu for “Crystal Search by:” from [Energy] to [Powder Pattern]. With this setting, the crystal structures found from the search are evaluated based on the similarity of their PXRD patterns to the PXRD pattern of experimental structure. Finally, click of [PXRD File:], and select the “pamoic-acid-c3.xrd” file that was prepared earlier.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the pamoic-acid-c3.ini.
pamoic-acid-c3.ini file
CRYSTAL_SEARCH CSP_SEARCH=POWDER_PATTERN CSP_SPGP=(P21/C) CSP_AUS_MODE=RANDOM CSP_ROT_MODE=RANDOM CSP_MAX_CRYSTAL=1000 CRYSTAL_OPTIMIZATION=ALL
[CRYSTAL_SEARCH] indicates that a crystal structure search will be executed.
[CSP_SEARCH=POWDER_PATTERN] means that the crystal structures found in the search will be evaluated based on the similarity of their PXRD patterns to the PXRD pattern of experimental structure.
[CSP_SPGP=P21/C] specifies the use of the space group P21/c in the search.
[CSP_ROT_MODE=RANDOM] and [CSP_AUS_MODE=RANDOM] mean that the initial molecular orientation and position will be determined randomly.
[CSP_MAX_CRYSTAL=1000] sets the number of trial crystal structures to 1000.
[CRYSTAL_OPTIMIZATION=ALL] specifies that “ALL” crystal structure optimizations will be applied.
Store the three files of pamoic-acid-c3.mol, pamoic-acid-c3.ini, and pamoic-acid-c3.xrd in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par pamoic-acid-c3enter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
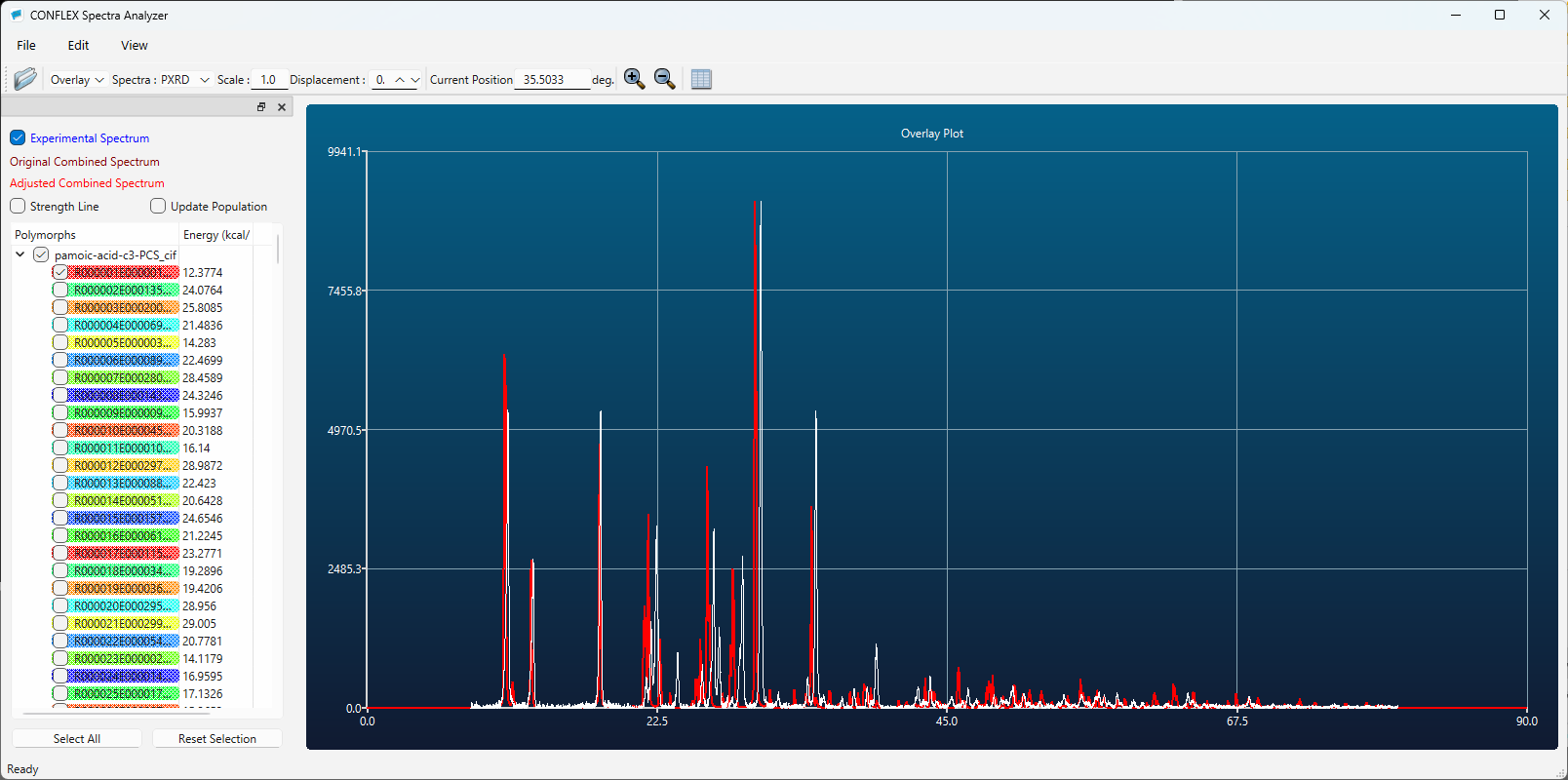
After the calculation, a file named pamoic-acid-c3.csp is generated, which provides detailed results of the structure search. When performing the search using reference diffraction data, the section titled [*** PREDICTED CRYSTAL STRUCTURES:] in the pamoic-acid.csp file lists the found crystal structures in order of similarity to the experimental PXRD pattern. The first structure in the list corresponds to the one with the highest similarity to the reference pattern. The structural data of the found crystals are saved in the pamoic-acid-c3-PCS.cif file. PXRD pattern similarity is evaluated usingGelder's method (R. de Gelder et al, J Comput Chem 22: 273–289, 2001.).
*** PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK R_RNK PXRD_SIM CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV
5 19 1 1 0.9056 12.3774 59.3730 -46.9957 1829.5937 1.4089 20.3676 4.9453 19.2844 90.0000 70.3767 90.0000 P21/C 225 10125 20.00 0
9 483 135 2 0.8351 24.0764 58.4811 -34.4047 2074.3915 1.2427 4.8375 19.5849 22.8573 90.0000 106.6807 90.0000 P21/C 188 8460 20.00 0
10 260 200 3 0.8276 25.8085 59.1800 -33.3715 1913.6902 1.3470 5.0699 25.0663 16.1916 90.0000 111.5615 90.0000 P21/C 204 9180 20.00 0
12 56 69 4 0.8235 21.4836 61.1121 -39.6285 1929.2047 1.3362 12.0076 12.5142 16.0878 90.0000 127.0568 90.0000 P21/C 205 9225 20.00 0
13 322 3 5 0.8219 14.2830 58.8049 -44.5220 1897.8000 1.3583 10.1471 4.8134 39.0029 90.0000 85.0212 90.0000 P21/C 202 9090 20.00 0
17 367 89 6 0.8182 22.4699 61.5496 -39.0797 1934.6148 1.3325 10.3329 20.6152 9.1642 90.0000 82.3239 90.0000 P21/C 197 8865 20.00 0
18 60 280 7 0.8177 28.4589 58.0370 -29.5781 1981.9578 1.3006 4.7838 11.1176 37.2683 90.0000 90.6615 90.0000 P21/C 202 9090 20.00 0
19 541 143 8 0.8137 24.3246 58.3375 -34.0128 1917.4130 1.3444 4.9060 15.6069 33.7231 90.0000 47.9523 90.0000 P21/C 202 9090 20.00 0
23 20 9 9 0.8133 15.9937 59.4918 -43.4981 1888.4289 1.3650 10.3216 9.1532 22.6717 90.0000 61.8423 90.0000 P21/C 197 8865 20.00 0
24 5 45 10 0.8122 20.3188 58.3402 -38.0214 1945.9817 1.3247 4.8345 10.1697 39.5911 90.0000 88.6467 90.0000 P21/C 204 9180 20.00 0
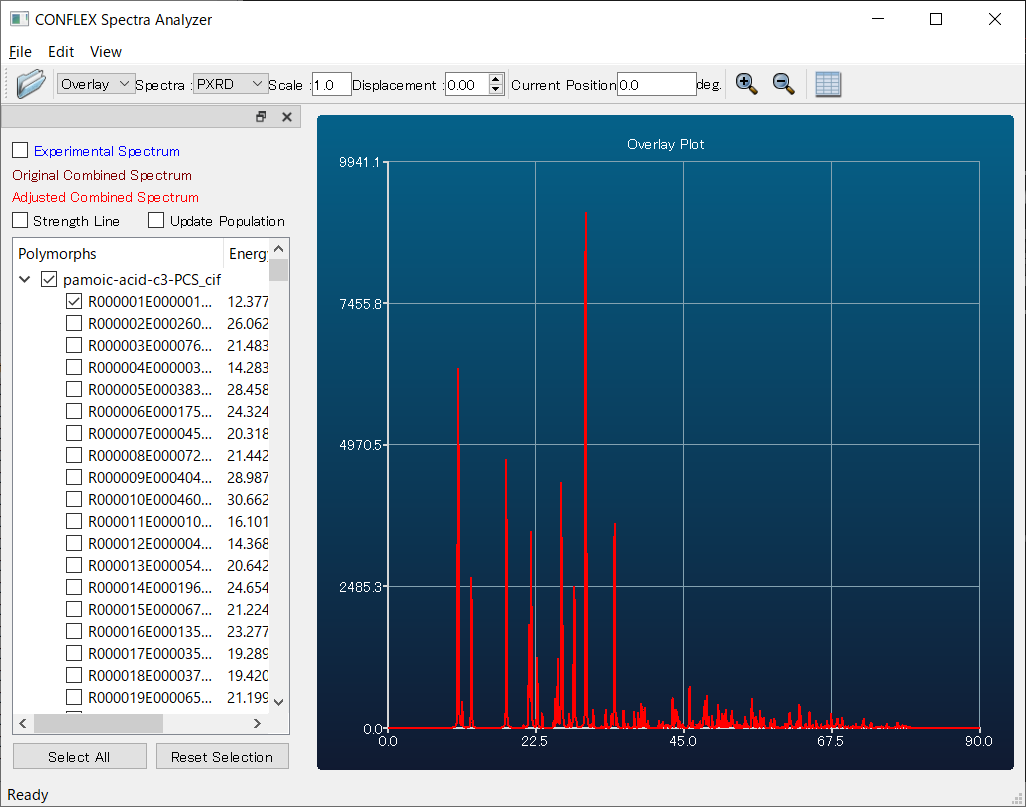
Here, by comparing the diffraction patterns of the first predicted structure (red) and the experimental one (white), you can see that they match well. Therefore, using the first structure as a starting model is expected to make structure refinement via Rietveld analysis much easier compared to performing the refinement without the support of CONFLEX.
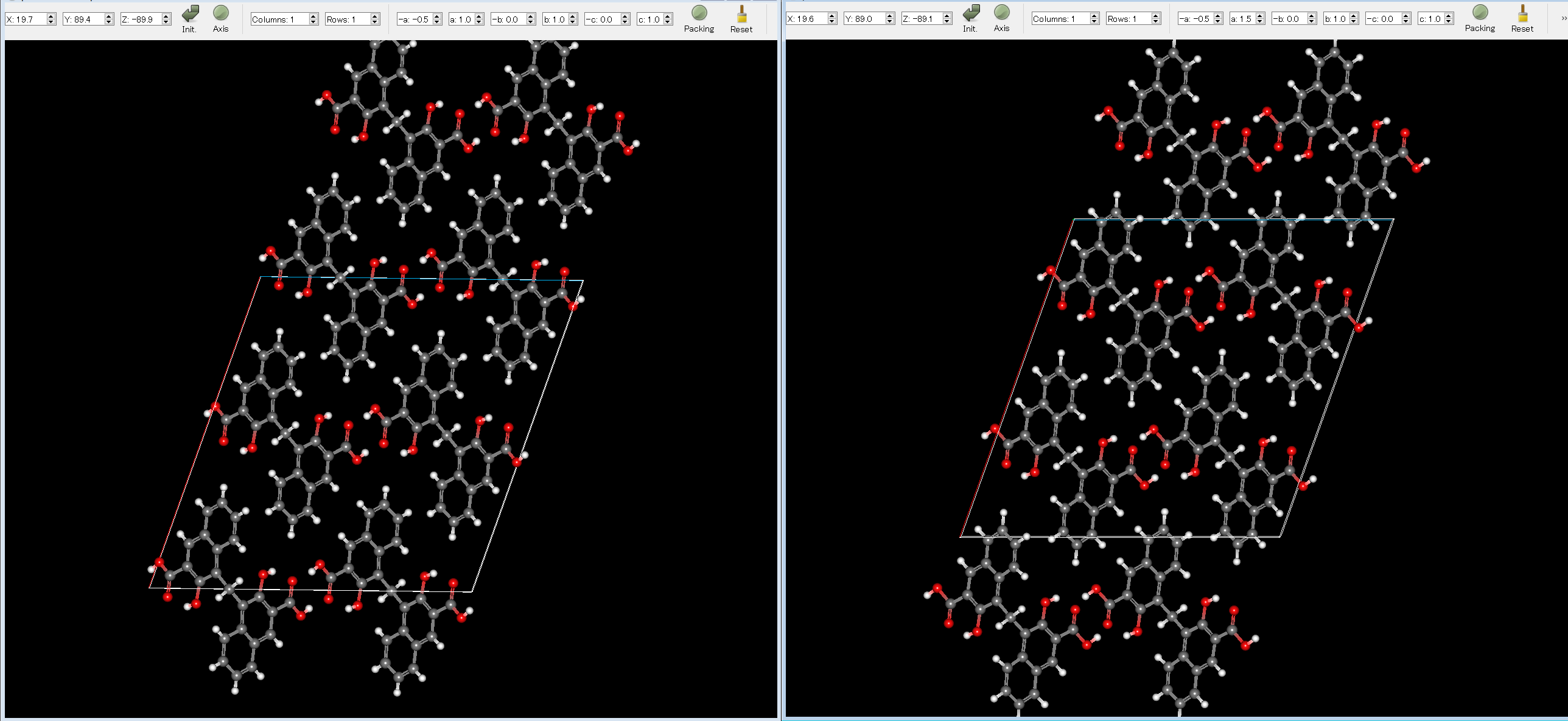
The experimental structure (left) and the first structure (right) are shown below. As you can see, the two structures match well.
[Output files]
After the crystal structure search calculation, the following files are generated.
| File type | Explanation |
|---|---|
| (Input file name).csp | Detailed information about the crystal structure search calculation is provided in this file. |
| (Input file name).cpt | This file contains the information needed to restart the calculation. |
| (Input file name).ical | Powder X-ray diffraction (PXRD) data for the crystal structures identified in the search are provided in this file. The corresponding crystal structure data are described in the -PCS.cif file. |
| (Input file name)-PCS.cif | The crystal structures identified in the search are provided in this file, ordered by crystal energy (or by similarity of PXRD patterns). |
| (Input file name)-FCS.cif | When the optimization of a trial structure is completed during the search process, the optimized structure is saved to this file. This allows you to view the optimized structure even before the entire search calculation is finished. |
In the [*** PREDICTED CRYSTAL STRUCTURES:] section of the csp file, the computationally suggested polymorphs are displayed in order of crystal energy.
*** PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV
4 318 1 -15.7921 4.6942 -20.4864 607.6657 1.3663 9.5996 8.3419 10.7545 90.0000 44.8779 90.0000 P21/C 365 4015 20.00 0
30 5295 2 -15.6634 4.7074 -20.3708 306.9963 1.3522 13.4774 7.0951 4.3469 90.0000 132.3915 90.0000 P21 371 4081 20.00 0
147 3622 3 -15.6125 4.7219 -20.3344 614.2783 1.3515 4.3718 19.8181 7.0899 90.0000 90.0000 90.0000 P212121 373 4103 20.00 0
167 3853 4 -15.6061 4.6925 -20.2986 612.8432 1.3547 9.6310 8.3434 7.6267 90.0000 90.0000 90.0000 P212121 325 3575 20.00 0
178 51 5 -15.5986 4.7162 -20.3147 614.9313 1.3501 4.3234 7.1069 21.9413 90.0000 114.1992 90.0000 P21/C 371 4081 20.00 0
190 90 6 -15.4618 4.7170 -20.1789 618.7977 1.3417 4.3746 7.0720 24.0800 90.0000 123.8359 90.0000 P21/C 374 4114 20.00 0
195 1948 7 -15.3029 4.7145 -20.0173 1222.4436 1.3583 14.5999 8.3633 10.4384 90.0000 73.5604 90.0000 C2/C 357 3927 20.00 0
206 169 8 -15.2948 4.7210 -20.0158 612.9536 1.3545 4.0230 8.3785 19.4984 90.0000 111.1490 90.0000 P21/C 358 3938 20.00 0
229 18 9 -15.2940 4.7263 -20.0203 612.5302 1.3554 8.3759 18.2972 8.8552 90.0000 26.8303 90.0000 P21/C 365 4015 20.00 0
290 4 10 -15.2939 4.7218 -20.0156 612.7425 1.3549 8.3793 18.2092 9.2919 90.0000 25.6065 90.0000 P21/C 357 3927 20.00 0
On the other hand, when the crystal structure search is performed using reference diffraction data, the results are listed in order of PXRD pattern similarity.
*** PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK R_RNK PXRD_SIM CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV
5 19 1 1 0.9056 12.3774 59.3730 -46.9957 1829.5937 1.4089 20.3676 4.9453 19.2844 90.0000 70.3767 90.0000 P21/C 225 10125 20.00 0
9 483 135 2 0.8351 24.0764 58.4811 -34.4047 2074.3915 1.2427 4.8375 19.5849 22.8573 90.0000 106.6807 90.0000 P21/C 188 8460 20.00 0
10 260 200 3 0.8276 25.8085 59.1800 -33.3715 1913.6902 1.3470 5.0699 25.0663 16.1916 90.0000 111.5615 90.0000 P21/C 204 9180 20.00 0
12 56 69 4 0.8235 21.4836 61.1121 -39.6285 1929.2047 1.3362 12.0076 12.5142 16.0878 90.0000 127.0568 90.0000 P21/C 205 9225 20.00 0
13 322 3 5 0.8219 14.2830 58.8049 -44.5220 1897.8000 1.3583 10.1471 4.8134 39.0029 90.0000 85.0212 90.0000 P21/C 202 9090 20.00 0
17 367 89 6 0.8182 22.4699 61.5496 -39.0797 1934.6148 1.3325 10.3329 20.6152 9.1642 90.0000 82.3239 90.0000 P21/C 197 8865 20.00 0
18 60 280 7 0.8177 28.4589 58.0370 -29.5781 1981.9578 1.3006 4.7838 11.1176 37.2683 90.0000 90.6615 90.0000 P21/C 202 9090 20.00 0
19 541 143 8 0.8137 24.3246 58.3375 -34.0128 1917.4130 1.3444 4.9060 15.6069 33.7231 90.0000 47.9523 90.0000 P21/C 202 9090 20.00 0
23 20 9 9 0.8133 15.9937 59.4918 -43.4981 1888.4289 1.3650 10.3216 9.1532 22.6717 90.0000 61.8423 90.0000 P21/C 197 8865 20.00 0
24 5 45 10 0.8122 20.3188 58.3402 -38.0214 1945.9817 1.3247 4.8345 10.1697 39.5911 90.0000 88.6467 90.0000 P21/C 204 9180 20.00 0
The meanings of the items in the [*** PREDICTED CRYSTAL STRUCTURES:] section are shown below.
| Item | Explanation |
|---|---|
| E_RNK | Ranking based on energy |
| R_RNK | Ranking based on PXRD pattern similarity |
| CRYST | Ecrystal |
| INTRA | Eintra |
| INTER | Elattice |
| VOL | Volume of unit cell |
| DES | Density of unit cell |
| A,B,C,ALPHA,BETA,GAMMA | Lattice constants |
| SPGP | Space group |
| NCALMOL | Total number of molecules included in the calculation |
| NCALATM | Total number of atoms included in the calculation |
| DMAX | Cut-off distance |
| NNEV | The number of negative eigenvalues |
* When different cut-off distances are used for vdW and Coulombic interactions, the values of “NCALMOL”, “NCALATM”, and “DMAX” correspond to the calculation of vadW interactions.
The ical file contains the powder X-ray diffraction data for the crystal structures found in the search. The -PCS.cif file contains the crystal structure data corresponding to the PXRD data. The “data_” in the -PCS.cif file and “NAME:” in the ical file correspond to the same structure.
------------ SIMULATED POWDER PATTERNS ------------
CID: 882
NAME: R000001E000001C000882
X-RAY: CO (KA1)
WAVE: 1.78899600
2*THETA: 8.010 - 79.990 , 0.010 STEP
H K L 2*THETA INTENSITY d
(DEGREE) (ANGSTROME)
0 0 0 8.010 1.167 0.00000
0 0 0 8.020 1.174 0.00000
0 0 0 8.030 1.182 0.00000
0 0 0 8.040 1.190 0.00000
0 0 0 8.050 1.197 0.00000
0 0 0 8.060 1.205 0.00000
0 0 0 8.070 1.213 0.00000
0 0 0 8.080 1.221 0.00000
0 0 0 8.090 1.229 0.00000
0 0 0 8.100 1.237 0.00000
0 0 0 8.110 1.246 0.00000
0 0 0 8.120 1.254 0.00000
0 0 0 8.130 1.263 0.00000
0 0 0 8.140 1.271 0.00000
* snip *
[Visualization of search results]
The polymorphs found in the search are saved in the -PCS.cif file. By opening the -PCS.cif file using the CONFLEX Interface, you can view them.
Select [Spectra_Analyzer] from the [Applications] menu with the -PCS file open, and you can also view the PXRD pattern of each structure.
[Restart of crystal structure search]
[Execution from Interface]
Store the molecular structure, ini, and cpt files used in the search calculation in a single folder. If reference diffraction data was used, the xrd file is also required.
Open the molecular structure file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
To restart the search calculation, check the [Restart calculation] checkbox at the bottom of the [Crystal Search] dialog in the detailed settings dialog.
The other settings are automatically loaded from the ini file. When completing the restart settings, click . If you added keywords manually, you should click and add the same keywords when restarting. If the settings for the search calculation do not match, the restart may not be performed correctly.
[Execution from command line]
Store the molecular structure, ini, and cpt files used in the search calculation in a single folder. If reference diffraction data was used, the xrd file is also required. Next, change the extension of the cpt file to “rst” and add the “CSP_RESTART” keyword to the ini file. Note that you should not change the settings for the search calculation in the ini file. If the settings do not match, the restart may not be performed correctly.
When you are ready to restart, execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par (Input file name)enter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
[Unavailable space groups]
The following space group can not be used in crystal structure search.
P4MM, P4BM, P42CM, P42NM, I4MM, I4CM, P-42M, P-421M, I-42M, P4/MMM, P4/NBM, P4/MBM,
P4/NMM, P42/MCM, P42/NNM, P42/MNM, P42/NCM, I4MMM, I4/MCM, P3, P31, P32, R3, P-3,
R-3, P312, P321, P3112, P3121, P3212, P3221, R32, P3M1, P31M, P3C1, P31C, R3M, R3C,
P-31M, P-31C, P-3M1, P-3C1, R-3M, R-3C, P6, P61, P65, P62, P64, P63, P-6, P6/M,
P63/M, P622, P6122, P6522, P6222, P6422, P6322, P6MM, P6CC, P63CM, P63MC, P-6M2,
P-6C2, P-62M, P-62C, P6/MMM, P6/MCC, P63/MCM, P63/MMC, P23, F23, I23, P213, I213,
PM-3, PN-3, FM-3, FD-3, IM-3, PA-3, IA-3, P432, P4232, F432, F4132, I432, P4332,
P4132, I4132, P-43M, F-43M, I-43M, P-43N, F-43C, I-43D, PM-3M, PN-3N, PM-3N, PN-3M,
FM-3M, FM-3C, FD-3M, FD-3C, IM-3M, IA-3D