- Products
- CONFLEX DOCK
CONFLEX DOCK is a docking simulation program that predicts where a specified peptide chain will coordinate and form a complex with a target protein.
Peptides are not only molecules that function directly as therapeutic agents, such as insulin, but are also known to mediate many protein-protein interactions inside cells. Additionally, peptides are attracting attention as materials in the field of medium-sized molecule drug discovery, due to advantages such as ease of synthesis and cell permeability.
By analyzing the details of protein-peptide interactions, valuable information can be obtained for designing effective drug molecules targeting specific proteins.
In CONFLEX DOCK, amino acid residues of the protein are coarse-grained using representative points (Cα atoms), and tetrahedra are constructed via Delaunay triangulation.
Docking poses are determined based on search points on the protein surface, which are arranged using a surface area calculation method commonly used for evaluating solvent effects. The peptide residues are placed on these search points on the protein surface. For affinity evaluation, a four-body potential, derived from experimental structure databases and using coarse-grained amino acid residues, is used as the scoring function.
While conventional programs usually calculate scores based on atomic interactions, using a coarse-grained potential allows for easier scoring and enables high-speed, comprehensive searches even for large proteins.
In other programs, the three-dimensional structure of the peptide is sometimes required as input data. However, CONFLEX DOCK only requires the three-dimensional structure of the protein and the sequence of the peptide.
If the structure of the peptide is known, its coordinate data can also be input to compare and evaluate differences from the search points.
For more details, please refer to the algorithm page.
Calculation Example
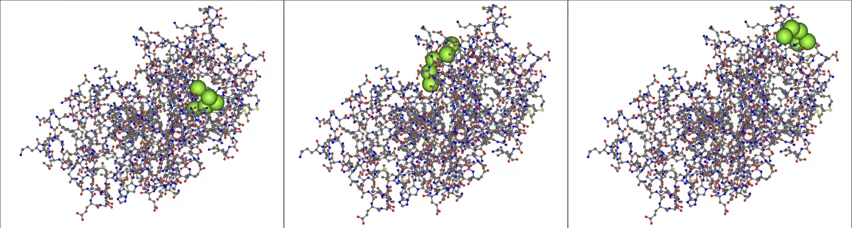
Below is an example of docking poses for a 5-residue peptide (Arg-Arg-Asp-Tyr-Phe, shown as green spheres) with Krev interaction trapped protein 1 (KRIT1, chain A of 4hdq.pdb). The scores for the poses, from left to right, are:
- 45.96
- 41.37
- 38.32
Reference
When publishing calculation results obtained with CONFLEX DOCK in a paper or other publication, please cite the following references:
T. Yamamoto, Y. Ikabata, H. Goto,
“Reconstruction of Four-Body Statistical Pseudopotential for Protein-Peptide Docking”,
J. Comput. Chem., Jpn.-Int. Ed., 2024, 10, 2023-0039.