- Products
- Work with Gaussian
CONFLEX optimize geometry or search conformations using classical molecular force field, normally. CONFLEX version 8 and later can invoke external Gaussian 09 or 16 program to optimize geometry, also. With this new feature, CONFLEX can handle molecules lacking force field parameters and/or atom types, such as metal complex, etc...
CONFLEX invoking Gaussian
The example of conformation searching using Gaussian is provided here.
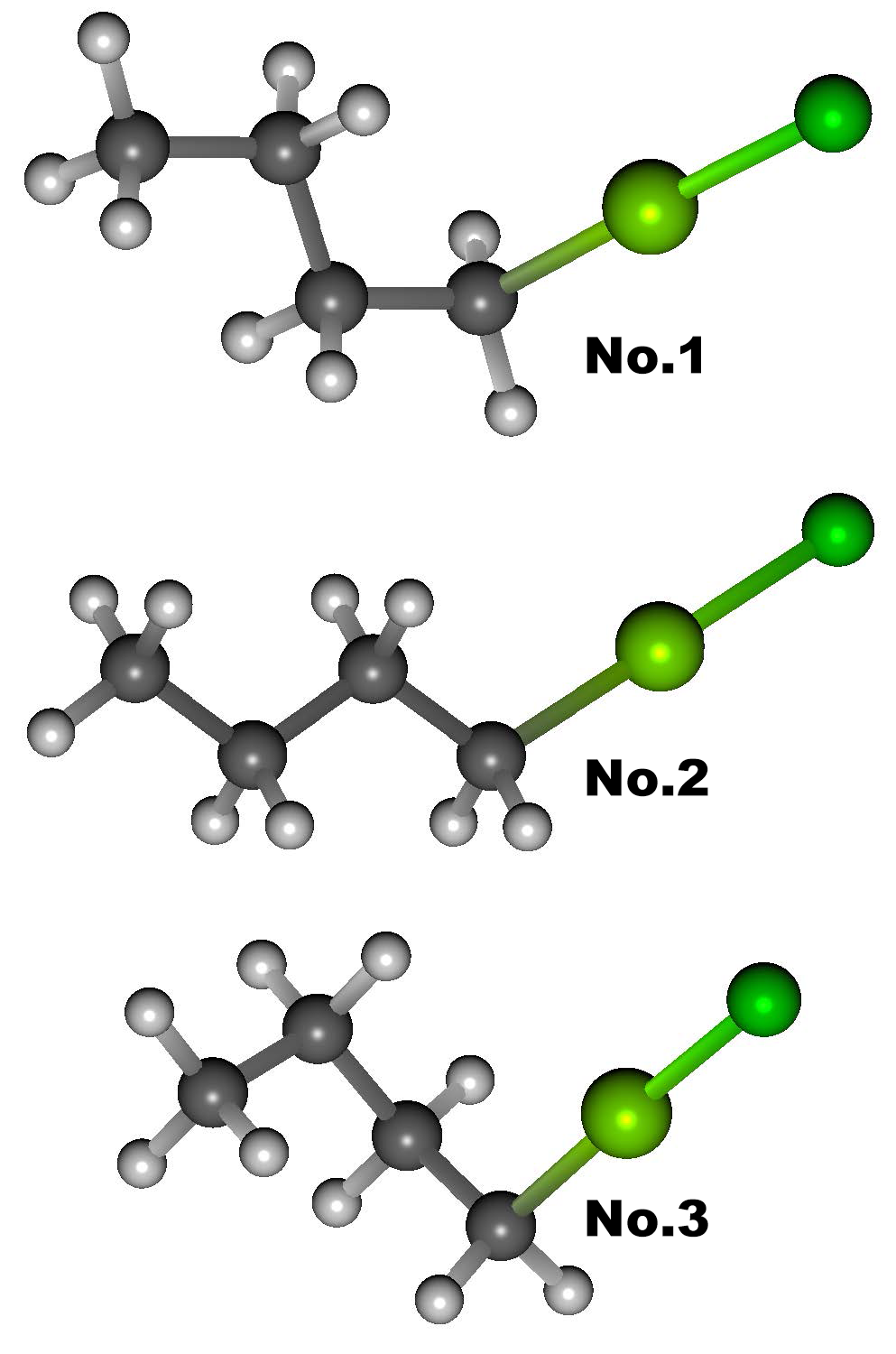
A target molecule is a Grignard reagent: Butyl Magnesium Chloride [(CH3(CH2)3MgCl].
Default MMFF94s force field does not include Mg atom with two covalent bonds. So, we used Gaussian with DFT method: B3LYP and 6-31G(d) basis set here. CONFLEX found 5 conformers (see table below). The pictures on the right shows Top 3 conformers found by CONFLEX and Gaussian.
| No. | Relative Energy (kcal/mol) |
|---|---|
| 1 | 0.00 |
| 2 | 0.19 |
| 3 | 0.77 |
| 4 | 1.00 |
| 5 | 1.13 |
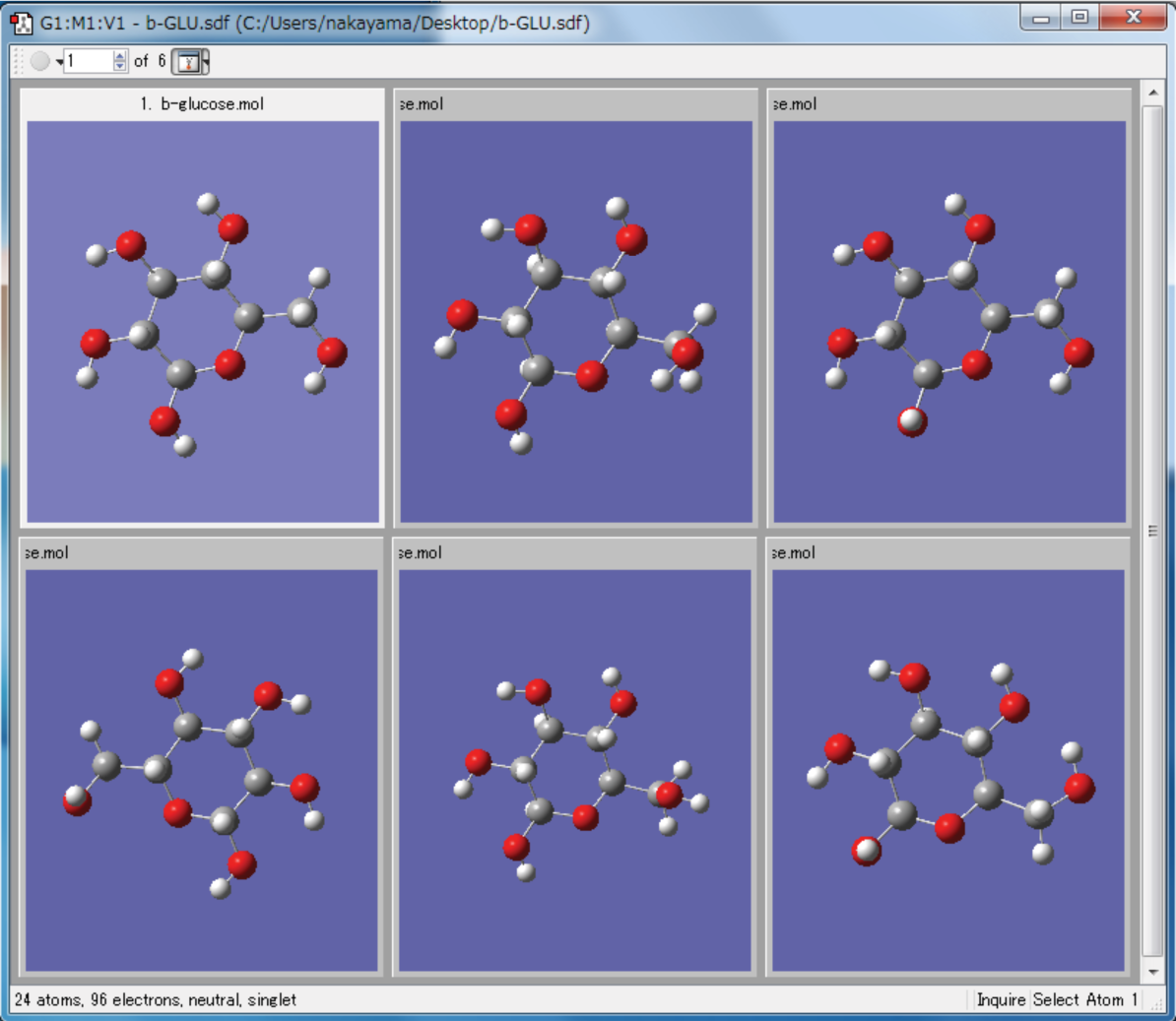
CONFLEX Output with GaussView
The conformation file generated by CONFLEX can be read with GaussView (see below).
GaussView can convert each conformers to Gaussian input files. User can simulate dipole moment, NMR chmical shifts and excitation energies etc. with these input files using Gaussian.