- 製品&サービス
- CONFLEX DOCK
CONFLEX DOCKは、基質となるタンパク質に対して、指定したペプチド鎖がどこに配位し複合体を形成するかを予測するドッキングシミュレーションプログラムです。
細胞内に存在するタンパク質にとってペプチドは、インスリン等のように治療薬として直接機能する分子が存在するだけでなく、多くのタンパク質間相互作用を媒介する役割を果たしていることが知られています。またペプチドは、合成の容易さや細胞内への浸透性等の利点があることから、中分子創薬の分野でも注目されている素材の一つです。
タンパク質-ペプチド間の相互作用の詳細を解析することで、標的タンパク質に対して有効な薬剤分子を設計するための有用な情報が得られることが期待できます。
CONFLEX DOCKで予測する際には、タンパク質のアミノ酸残基を代表点(Cα原子)で粗視化し、ドロネー分割により四面体を構築します。
ドッキングポーズは、溶媒効果を求める際に用いられる表面積計算手法を適用して配置した、タンパク質表面上の探索点をもとに求めます。タンパク質表面に設定した探索点にペプチドの残基を置き、それらの親和性の評価のために、アミノ酸残基を粗視化して実験構造データベースをもとに求めた4体ポテンシャルをスコアとして用いています。
従来のプログラムでは、原子間相互作用をもとにスコアを算出することがほとんどですが、粗視化したポテンシャルを用いることでスコアの算出が容易になり、巨大なタンパク質でも高速かつ網羅的に探索することが可能です。
他のプログラムでは、入力データとしてペプチドの三次元構造が必要な場合がありますが、CONFLEX DOCKの計算に必要な情報はタンパク質の三次元構造とペプチドの配列のみです。
ただし、ペプチドの構造が既知である場合、その座標データを入力することで探索点との差を算出し、比較することも可能にしています。
計算例
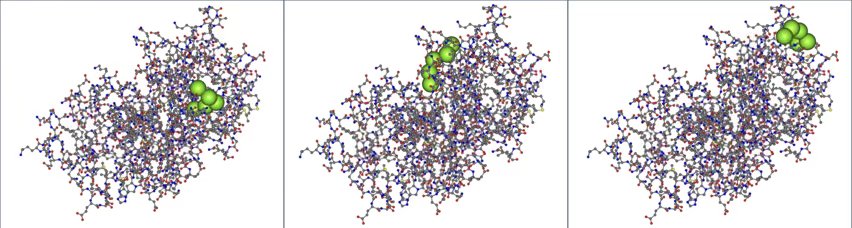
計算例として、Krev interaction trapped protein 1(KRIT1、4hdq.pdbのA鎖)に対する5残基のペプチド(Arg-Arg-Asp-Tyr-Phe、緑色の球)のドッキングポーズの一部を以下に示します。スコアは左からそれぞれ 45.96、41.37、38.32 です。
【文献引用】
CONFLEX DOCKで得られた計算結果を論文等に掲載する場合は、以下の文献を引用してください。
T. Yamamoto, Y. Ikabata, H. Goto,
“Reconstruction of Four-Body Statistical Pseudopotential for Protein-Peptide Docking”,
J. Comput. Chem., Jpn.-Int. Ed., 2024, 10, 2023-0039.