- 製品&サービス
- CONFLEX新機能
CONFLEX 9 新機能

- 結晶計算機能の追加(Pro) 分子間に調和ポテンシャルを適用することで、疑似的な力を付加できるようになりました。非対称単位の分子間配置に制約を課すことができます。
- 結合次数補正機能の追加(Basic, Pro) 結合の総数が原子価より少ない原子同士が結合している場合、水素原子を付加する前にその結合の次数を補正する機能を追加しました。(.pdbを除く)
- Gaussian呼出機能の拡張(Pro) 部分的に構造を固定するキーワードと同時に指定することで、固定箇所がGaussianの入力ファイルに反映されるようになりました。
- 動作環境
赤字がRev.Cより対応
Windows (64bit): 10, 11(21H2以降)
macOS: 12, 13 (Apple Siliconは13のみ)
Linux CONFLEX: CentOS 7.6-7.9, RedHat EL 8.4, Ubuntu 22.04
(他のLinuxへの対応についてはお問い合わせください)
また、Gaussianの入力ファイルに結合情報を含められるようになりました。
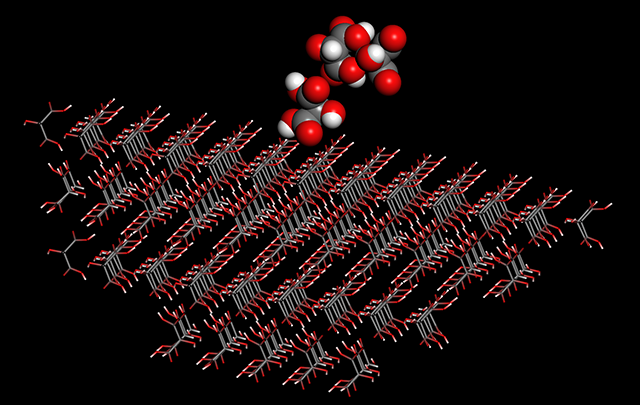
- 結晶表面解析機能の追加(Pro) 指定した結晶面の比表面エネルギーが計算できるようになりました。
- Distance Dependent Dielectric Constant の導入(Basic, Pro) 静電相互作用項に含まれる比誘電率が原子間距離に比例する計算手法を導入しました。
- 動作環境
赤字がRev.Bより対応
Windows (64bit) 10, 11
macOS (x86_64) 10.15, 11, 12
Linux CONFLEX: CentOS 7.6-7.9, RedHat EL 8.4
(他のLinuxへの対応についてはお問い合わせください)
結晶は複数の結晶面からなる多面体ですが、結晶面の比表面エネルギーはそれぞれ異なります。よく発達する結晶面の比表面エネルギーは小さいと考えられるため、その解析は結晶の外形や成長性などの研究に有効です。
定数で計算する従来の手法と比べて溶媒による寄与をより精度良く記述することができます。
- 結晶構造最適化の機能強化 結晶構造最適化に完全対角Newton-Raphson法が利用できるようになりました。結晶構造探索にかかる時間を大幅に削減できます。
- Ewald法の導入 静電相互作用計算にEwald法が利用できるようになりました。遠距離まで働く静電相互作用を高速かつ高精度に計算することができます。
- AMBER力場を用いた計算機能(構造最適化、配座探索のみ) AMBER用の入力ファイルを用いて、AMBER力場による構造最適化と配座探索を行えるようになりました。
- GB/SA計算のオプション追加 静電相互作用項のみ計算に取り入れるオプションを追加しました。
- MMFF94sパラメーターの一部修正 アミド基のN-H伸縮パラメーターを、元のMMFF94sの値に戻しました。
- 動作環境
赤字がCONFLEX 9 Rev.Aより対応
Windows 10
macOS 10.14, 10.15
Linux CentOS: 7.6-7.9, 8.0-8.3
(他のLinuxへの対応についてはお問い合わせください)
CONFLEX Interface 9 新機能
- 大きな分子の表示を高速化しました
- 分子のボール&スティックおよびワイヤーフレーム表示を高精細化しました
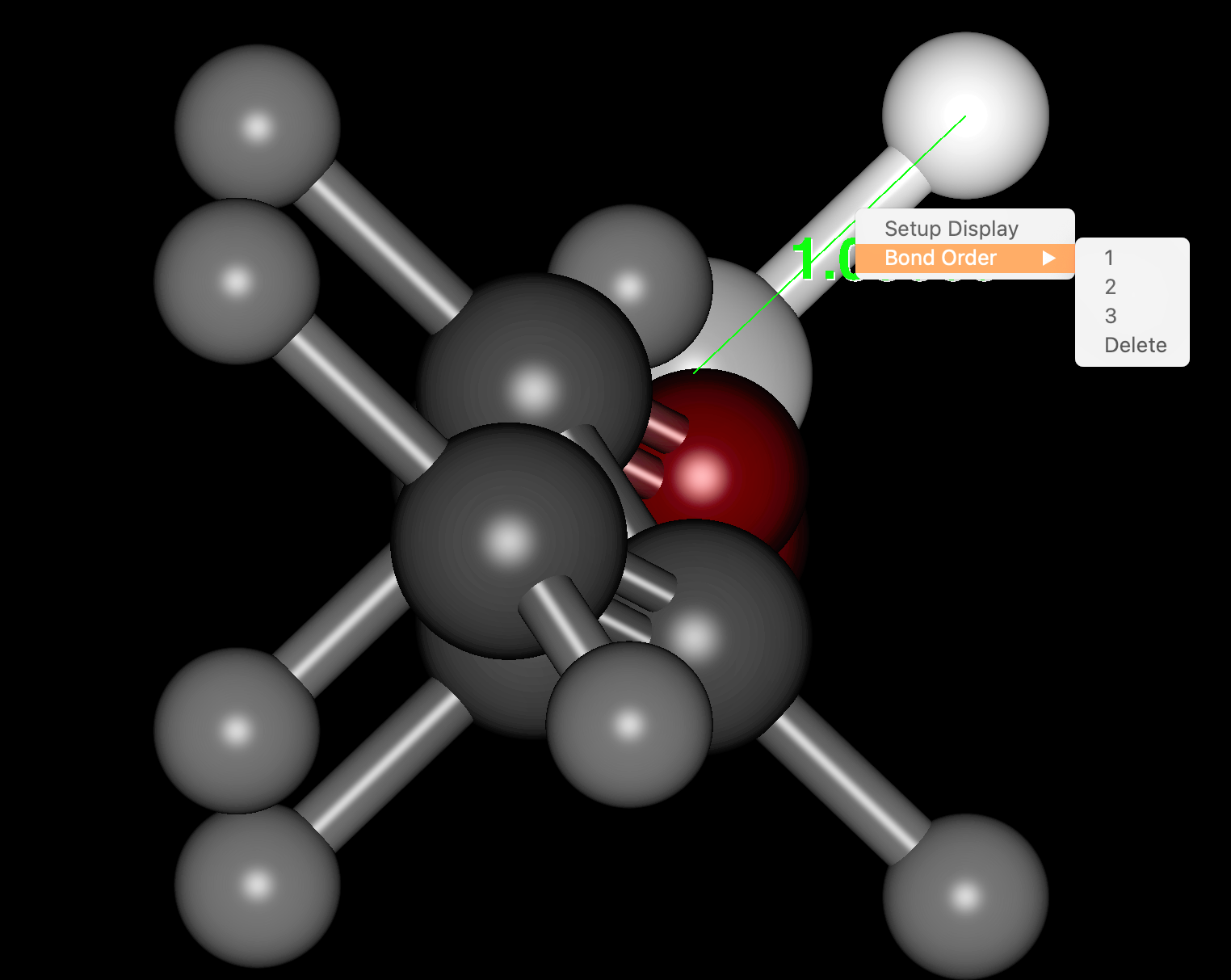
- 原子の形式電荷の変更が可能になりました(表示された原子上でマウスを右クリック)
- 結合多重度の変更が可能になりました(表示された結合上でマウスを右クリック)
- 動作環境
赤字がRev.Cより対応
Windows (64bit): 10, 11 (21H2以降)
macOS: 12, 13(Apple Siliconは13のみ)
Linux Interface: RedHat 8.4, Ubuntu 22.04
- CONFLEXの新機能に対応しました:
- 比表面エネルギーの計算設定と結果表示
- Distance Dependent Dielectric 計算の設定
- CONFLEX BSOファイルを読み込んだ際のサマリー表示を改良しました
- 多くの配座や結晶構造を含む、大きなサイズのファイルをバックグラウンドで読み込むようにしました
最初の構造が表示されるまでの待ち時間が短くなります - PDB, cifファイルの読み込みルーチンを改良しました
これまで読み込めなかったファイルの読み込みエラーが減少しています -
動作環境
赤字がRev.Bより対応
Windows (64bit) 10, 11 (21H2以降)
macOS (x86_64) 10.15, 11, 12
Linux RedHat EL 8.4
- 分子の一部の表示形態を変更できるようになりました
- 分割画面の各エリアの表示形態を個別に指定できるようになりました
- 原子の表示半径に任意の値を指定できるようになりました
- Windows 10に標準で組み込まれたOpenSSHに対応しました
- ワイヤーフレーム表示の際の線幅を指定できるようになりました
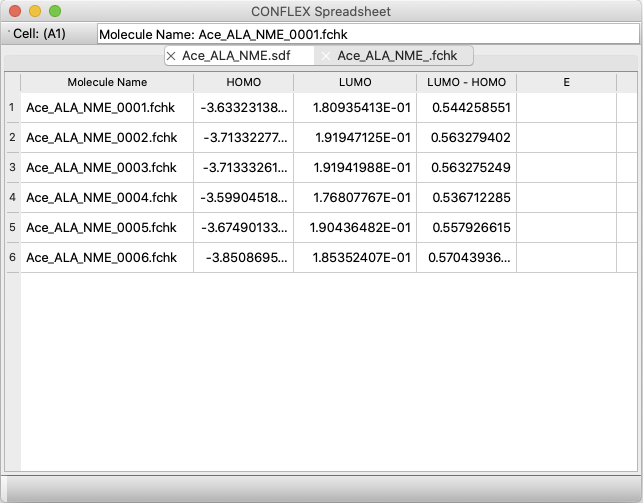
- 新アプリケーション「CONFLEX Spreadsheet」が追加されました:
- 配座ファイルを読み込み、計算プロパティを表形式に自動入力
- 結晶探索ファイルを読み込み、計算プロパティを表形式に自動入力
- 複数のGaussian FChkファイルを読み込み、計算プロパティを表形式に自動入力
- 複数のセルのコピー&ペースト
- Microsoft Excel形式での保存
- セル内の数値に対する、演算機能
- 動作環境
赤字がCONFLEX 9 Rev.Aより対応
Windows 10
macOS 10.14, 10.15
Linux CentOS: 7.6
CONFLEX 8 新機能
- NMR 3J結合定数のパラメーター設定
- Karplus式をベースに、任意の原子間での3J結合定数を求めることが可能です。
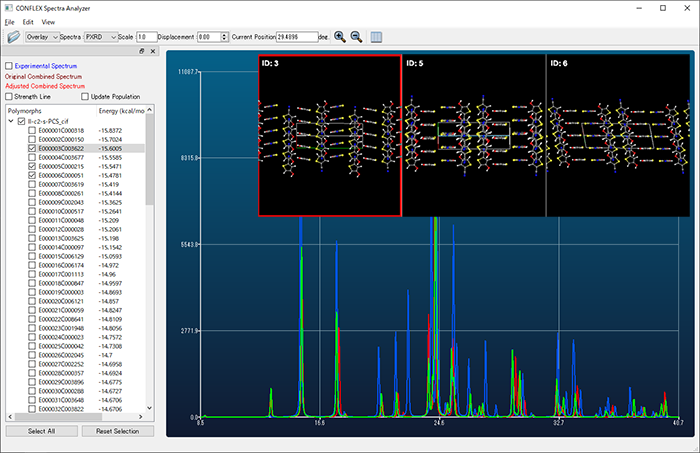
- 分子性結晶構造のクラスタリング
- 結晶構造探索により得られた構造を、粉末X線回折パターンの類似度に基づいて、グループ分け出来るようになりました。実測値に対応した結晶構造群を見出すことが容易になります。
- 結晶構造最適化の機能強化
- 指定した分子の構造・位置を固定しながら、
結晶構造最適化ができるようになりました。
- 指定した分子の構造・位置を固定しながら、
- PPPパラメーター設定
- 動作環境
-
赤字がCONFLEX 8 Rev.Cより対応
Windows・CONFLEX&Interface: 8.1, 10
macOS・CONFLEX&Interface 10.13, 10.14
Linux・CONFLEX: CentOS 6.6-6.10, 7.2-7.6, 8.0
Linux・Interface: CentOS 7.2-7.6
- 部分的に構造を固定した構造最適化、配座探索の実装
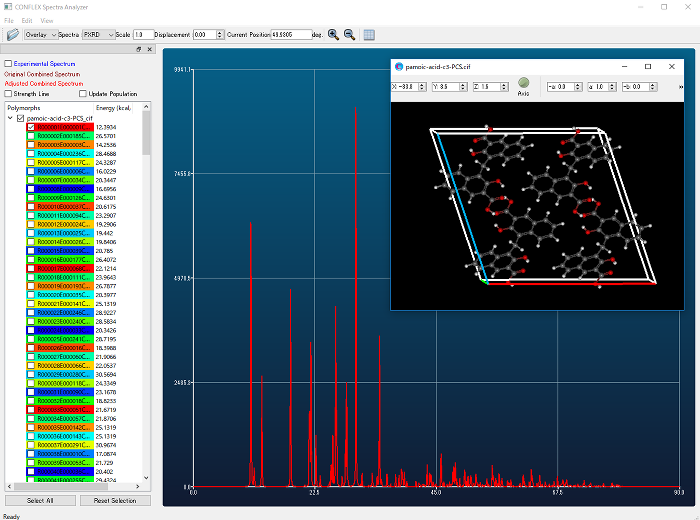
- 構造最適化の際に、座標を固定するか最適化するかを原子ごとに指定できるようになりました。
- 配座探索と組み合わせてお使い頂くことも可能です。
- 粉末X線回折スペクトル類似度評価法の導入
- 結晶構造探索により得られた構造の粉末X線回折スペクトルを、実測データとの類似度で評価できるようになりました。
- 動作環境
-
赤字がCONFLEX 8 Rev.Bより対応
Windows・CONFLEX&Interface: 7, 8.1, 10
macOS・CONFLEX&Interface 10.12, 10.13
Linux・CONFLEX: CentOS 6.6-6.9, 7.2-7.5
Linux・Interface: CentOS CentOS 6.6, 7.2
(他のLinuxへの対応についてはお問い合わせください)
- 動的反応座標(Dynamic Reaction Coordinate, DRC)の導入
- 基準振動モードを使用して初期速度ベクトルを算出し、動力学計算を行う計算方法です。
- 複数分子の配置変換や、大きな分子の配座変換に適用できます。
- 構造最適化および配座探索時の外部プログラム(Gaussian 09,16)呼び出し機能
- 同じマシンにGaussianがインストールされている場合、CONFLEXからGaussianを呼び出して構造最適化および配座探索計算を直接行うことができます。
- 分子力場パラメーターの無い分子や、古典力場では扱えない電子状態での構造最適化・配座探索が可能です。
- CONFLEXの探索アルゴリズムをそのまま用いながら、設定した電子状態計算レベルによる構造最適化を行い得られたエネルギー値で配座異性体を評価出来ます。
-
結晶構造最適化計算の共有メモリー型並列処理(OpenMP)による高効率化(Parallel CONFLEXのみ)
- 結晶構造を最適化する際の分子間相互作用計算を、OpenMPによる並列処理で高速化しています。
- OpenMP化コードの導入に伴い、結晶構造最適化におけるMPI並列機能は廃止します。
-
結晶構造探索計算のMPI/OpenMPハイブリッド計算の実現 (Parallel CONFLEXのみ)
- 結晶構造探索時のMPI並列処理と、個々の結晶構造最適化計算でのOpenMP並列処理を、同時に行えるようになりました。
- 保存する配座を二面角で決定(CHECK=TORSION)する際の処理の高速化
- 計算に用いる元素質量の改訂
- 以下の参考文献に掲載されている元素質量に変更しています。
B. Pfeiffer, K. Venkataramaniah, U. Czok, C. Scheidenberger, "Atomic mass compilation 2012", Atomic Data and Nuclear Data Tables, 2014, 100, 403-535.
- 以下の参考文献に掲載されている元素質量に変更しています。
- 動作環境
- 以下のOSに対応しています。(赤字がCONFLEX 8 Rev.Aより対応)
Windows: 7, 8.1, 10
Mac: macOS 10.11, 10.12 (10.13への対応についてはお問い合わせください)
Linux: CentOS 6.6-6.9, 7.2-7.4 (他のLinuxへの対応についてはお問い合わせください)
- 以下のOSに対応しています。(赤字がCONFLEX 8 Rev.Aより対応)
CONFLEX Interface 8 新機能
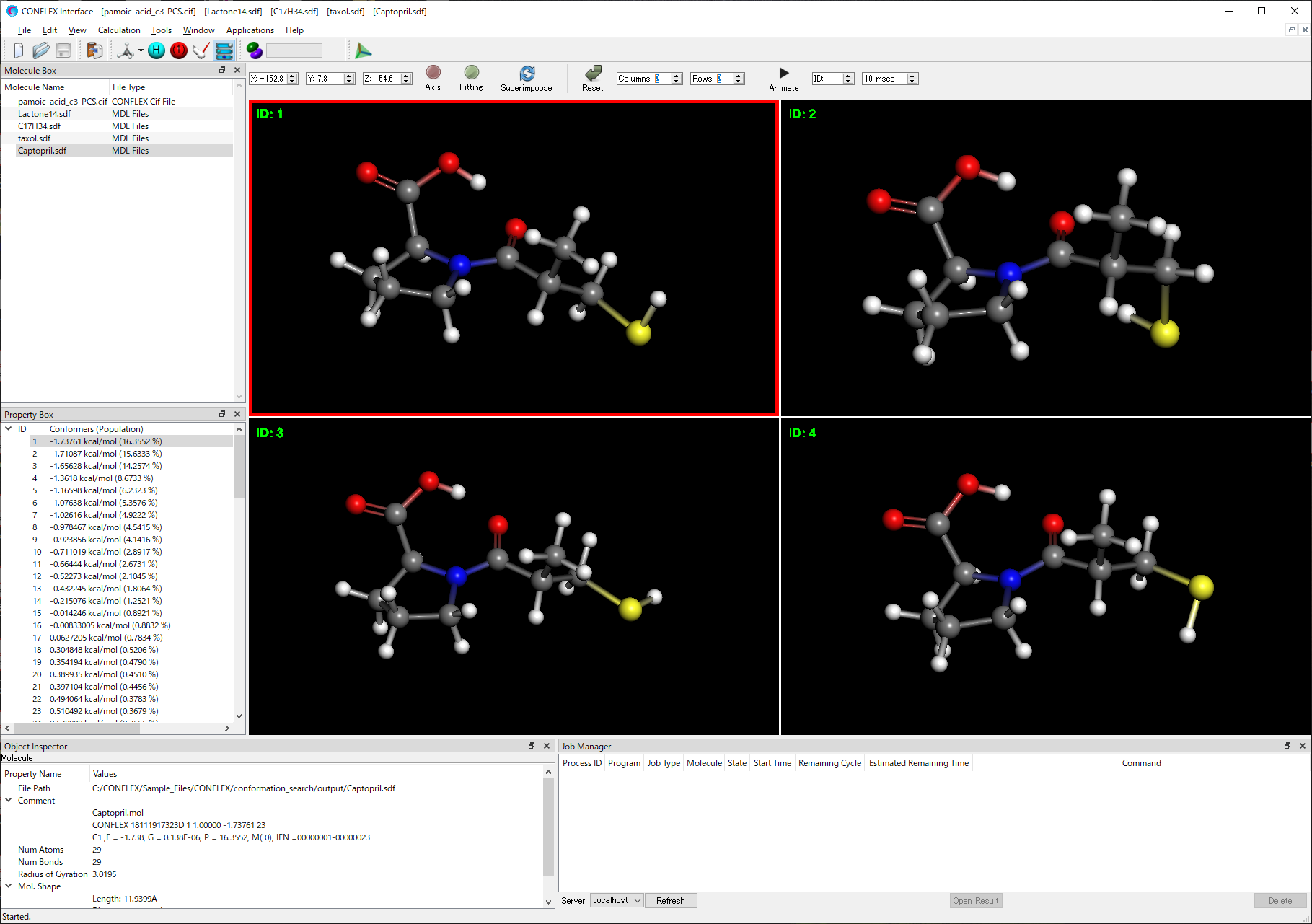
- 画面を分割し、それぞれに別の配座や結晶構造を表示
- 複数配座の、指定した原子での重ね合わせ表示
- プロパティダイアログ内の表示がソート可能
- DRC計算結果の動画表示
- 大量の構造を含む結晶探索結果ファイルの読み込みの高速化
-
GPUによる高速化
※OpenGL 3.3以上が必要です。
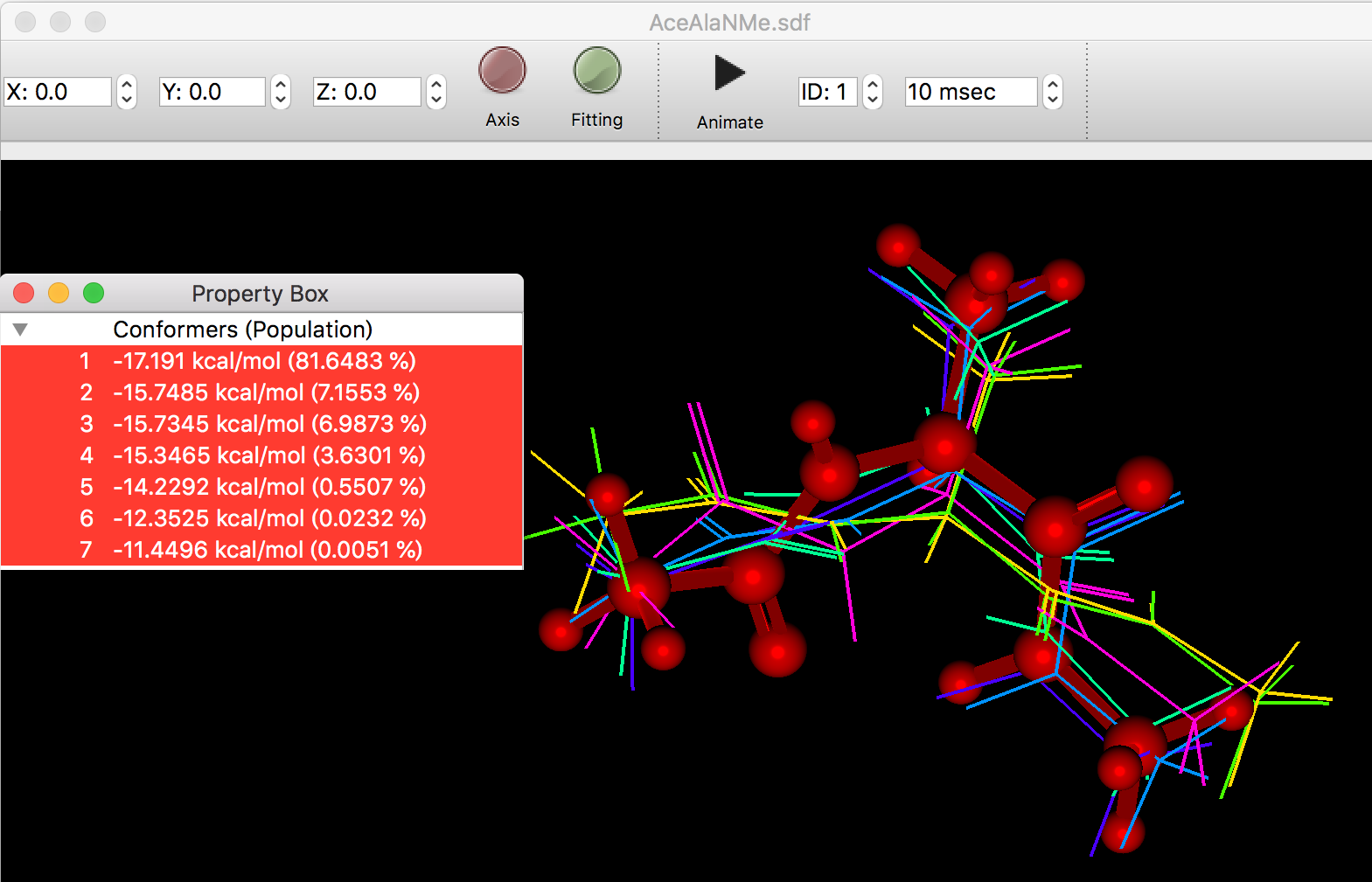
- 配座のフィッティングおよび重ね合わせ機能の追加
- PBS Pro に対応:サーバーへのジョブ投入時に利用可能
- GPU を利用した高速化(主に原子ラベル関連)
- スペクトル解析機能:Spectra Analyzer の機能強化
- ズームレベルに応じて、スペクトル表示の解像度の最適化
- 粉末X 線回折スペクトル表示への対応
- 粉末X 線回折スペクトル表示への実験データの読み込み機能
- 希望のピークに対するラベル付け機能
- グラフィック表示の強化
- OpenGL SLへの対応による高速化
- 振動モードのアニメーション表示
- DRC計算結果のムービー表示
- 結晶面をh, k, lで指定して表示
- 分子のライン表示オプション
- (Mac) Retinaディスプレイへの対応
- 新規ツールバーの搭載
- XYZ座標軸表示の切り替え
- 結晶のユニット・ボックス表示の切り替え
- 結晶表示の大きさの設定
- アニメーション表示の再生・停止・コマ送り戻し
- アニメーション・スピード
- 振動アニメーションの大きさの倍率
- 分子面表示の透明度の設定
- 分子面の大きさを規定する電子密度の設定
- 新規計算設定ダイアログ
- Host - Ligandサーチ
- DRC計算
- 外部プログラム呼び出し設定
- OpenMP/MPIパラレル計算設定
- 簡易結晶群選択ページ
- PDBファイル読み込みの強化
- アミノ酸・核酸を含むPDBファイルを読み込む際に標準構造から欠落している原子を認識&補完
- 原子間距離・結合角・ねじれ角ラベル
- 構造ラベルのWatchリスト機能
- 結合していない原子間での原子間距離・結合角・ねじれ角ラベル
- 結晶表示中の分子間ラベル表示
- ラベルの複数表示
- プログラムの設定ファイルの保存場所を変更
- セキュリティーの問題から、レジストリーやアプリケーション内部の変更をやめ、以下の場所に変更
- Win: FOLDERID\_RoamingAppData/conflex.co.jp/CONFLEX.ini
- Unix: $HOME/.config/conflex.co.jp/CONFLEX.ini
- Mac: $HOME/.config/conflex.co.jp/CONFLEX.ini
- セキュリティーの問題から、レジストリーやアプリケーション内部の変更をやめ、以下の場所に変更
CONFLEX 7 新機能
- 結晶表面解析機能の実装
- 超球状結晶を設定したミラー指数で切断し、その切断面に最も近い分子、または切断面上にある分子が受ける分子間相互作用を計算します。
- 昇華エネルギーや吸着エネルギーなど、結晶表面に関わるエネルギーの解析に役立ちます。
- 結晶構造探索計算の並列化性能の向上 (Parallel CONFLEXのみ)
- 結晶構造探索で行っている結晶構造最適化計算を分散させることで、探索計算の並列化効率が飛躍的に上昇しました。
- 結晶エネルギーの定義の変更
- 格子エネルギーや結晶構造探索の精度向上のため、結晶エネルギーの定義を変更しました。
- mol2ファイルへの対応 (直接計算実行が可能)
- mol2ファイルを入力データとして構造最適化や配座探索を行い、mol2形式の出力ファイルを得ることが可能になりました。
- ねじれ相互作用の6次項までの拡張
- 通常の分子力場では3次または4次まで考慮されるねじれ相互作用について、6次項までパラメーターを設定して計算を行うことが可能になりました。
- 配座探索中のE-Z配置の制限を外すキーワードの追加
- 通常の配座探索では、探索中に二重結合周りのE-Z配置が反転した構造が得られた場合その構造を破棄しますが、これらを保存するためのキーワードを追加しました。
- 動作環境
- 以下のOSに対応しています。(赤字がRev.Dより対応)
Windows: 7, 8.1, 10
Mac: OSX 10.9, 10.10, 10.11
Linux: CentOS 6.5, 6.6, 6.7, 6.8 (その他のLinuxはご相談下さい)
- 以下のOSに対応しています。(赤字がRev.Dより対応)
- CONFLEX
- ホスト−リガンド配位探索機能の追加
- 複数の分子を含む系について、ある一つの分子が他の分子(群)に対してどの位置にどのような向きで配位するのが安定かを探索します。
- 二量体や錯体の安定構造の探索に利用出来ます。
- Parallel CONFLEX for Windows
- Windowsマシン上で、MPI並列計算を実行できるようになりました。
複数のCPUコアを有するWindowsマシンでの配座探索や分子性結晶計算が、より効率よく行えます。(Parallel CONFLEXのライセンスが必要です。)
- Windowsマシン上で、MPI並列計算を実行できるようになりました。
- ホスト−リガンド配位探索機能の追加
- Interface
- Windows上でのParallel CONFLEXの実行
- CONFLEX簡易実行ダイアログで、Search Limitの設定が可能に
- ローカルマシンでのGaussianジョブ実行に対応
- Gaussianジョブの簡易実行機能
- 簡易ジョブ実行内容の編集エディター
- Gaussianによるスペクトル計算結果の存在確率に沿った重ね合わせ
- Platform LSFのHostGroupの設定に対応
- 数値入力による分子回転
- 分子情報ダイアログに分子形状の情報を表示
- 動作環境
- 以下のOSに対応しています。(赤字がRev.Cより対応)
Windows: 7, 8.1 [32/64bit]
Mac: OSX: 10.8, 10.9, 10.10
Linux: CentOS 6.1-6.5, 6.6, 7.0、Ubuntu 12.04.3, 14.04
- 以下のOSに対応しています。(赤字がRev.Cより対応)
- CONFLEX
- ユーザー定義パラメーターの追加機能
- CONFLEXに含まれている力場パラメーターに対応していない原子タイプを含む分子について、パラメーターを追加して計算することができます。
- 既存のパラメーターを修正して計算することも可能です。
- MMFF94s力場のみの対応です。
- 圧力を考慮した結晶構造最適化機能(三斜晶系、単斜晶系、斜方晶系のみ)
- 結晶構造を最適化する際、外部から加わる圧力を指定して最適化することができます。
- 三斜晶系、単斜晶系、斜方晶系のみの対応です。
- ユーザー定義パラメーターの追加機能
- Interface
- 配座リスト(拡張子ls1のファイル)表示機能
- 等電子密度面の作成&表示機能の拡張
- ジョブスケジューラーPlatform LSFに対応
- 計算スキームの簡易編集機能
- 各種スペクトル表示機能
(振動スペクトル、UV/Visスペクトル、UV/CDスペクトル、プロトンNMRピーク形状)
- 動作環境
- 以下のOSに対応しています。(赤字がRev.Bより対応)
Windows: 7, 8.1 [32/64bit]
Mac: OSX 10.6, 10.7, 10.8, 10.9
Linux: CentOS 6.1-6.5, Ubuntu 12.04.3
- 以下のOSに対応しています。(赤字がRev.Bより対応)
- 結晶構造探索
- 分子構造データと空間群の対称性を入力することで、自動的に結晶構造を作成し、構造最適化を行ない、エネルギー極小に位置する結晶構造を網羅的に算出します。
- 最適化した一連の結晶構造に対して、エネルギーの低い順に並べるだけでなく、あらかじめ用意した粉末回折データに近い順に並べることもできます。
- 粉末X線回折データの出力
- 結晶構造の粉末回折データを算出し出力します。X線源の元素や波長を変えることも可能です。
- 新型CONFLEX Interface登場
- CONFLEX Interfaceが一新し、Windows、Mac、Linuxに対応しました。
- ネットワーク経由でのジョブの実行が可能になりました。例えば、Windows PCからLinuxサーバーへ計算を実行することが出来ます。またLinuxサーバー上で得られた結果をそのままWindows PCで読み込むことが出来ます(サーバーには、GridEngineのインストールが必要になります)。
- 外部プログラムとの連携が可能です。例えば、Gaussianのネットワーク経由での計算実行が出来ます(サーバーには、GridEngineのインストールが必要になります)。
- 以下のファイルの読み込みが可能です。
- MDLファイル:.mol .sdf
- CONFLEX出力ファイル:.bso .sdf
- Sybyl mol2 ファイル
- PDBファイル
- 結晶構造データファイル:.cif .cmf
- Gaussianチェックポイントファイル:.fchk
- GAMESSログファイル
- Fireflyログファイル
- ChemDrawからのカット&ペースト
- ChemOfficeシリーズに対応
-
ChemOfficeシリーズ v13にCONFLEXのインターフェイスが搭載されました。
ChemBioOffice Ultra v13、ChemOffice Pro v13、ChemBio3D Ultra v13のいずれかをご購入いただくと、直接CONFLEXの計算実行が可能です。
-
ChemOfficeシリーズ v13にCONFLEXのインターフェイスが搭載されました。